

Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7
- PMID: 30381411
- PMCID: PMC6411060
- DOI: 10.1126/scitranslmed.aap8677
Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7
Abstract
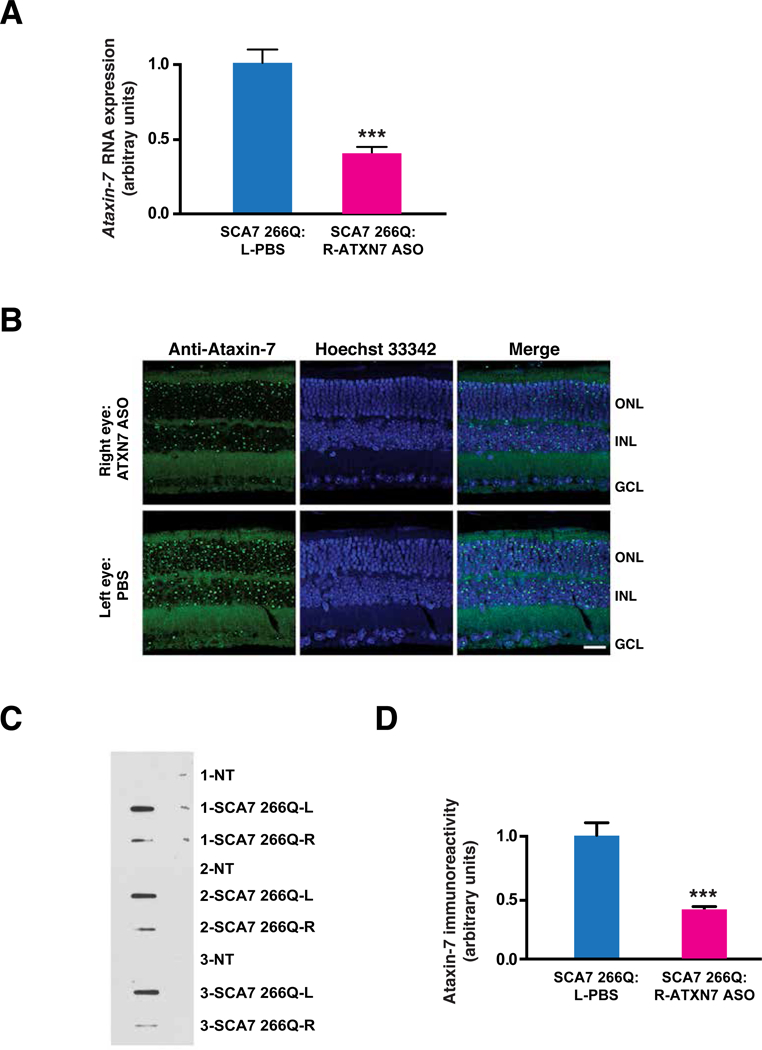
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant neurodegenerative disorder characterized by cerebellar and retinal degeneration, and is caused by a CAG-polyglutamine repeat expansion in the ATAXIN-7 gene. Patients with SCA7 develop progressive cone-rod dystrophy, typically resulting in blindness. Antisense oligonucleotides (ASOs) are single-stranded chemically modified nucleic acids designed to mediate the destruction, prevent the translation, or modify the processing of targeted RNAs. Here, we evaluated ASOs as treatments for SCA7 retinal degeneration in representative mouse models of the disease after injection into the vitreous humor of the eye. Using Ataxin-7 aggregation, visual function, retinal histopathology, gene expression, and epigenetic dysregulation as outcome measures, we found that ASO-mediated Ataxin-7 knockdown yielded improvements in treated SCA7 mice. In SCA7 mice with retinal disease, intravitreal injection of Ataxin-7 ASOs also improved visual function despite initiating treatment after symptom onset. Using color fundus photography and autofluorescence imaging, we also determined the nature of retinal degeneration in human SCA7 patients. We observed variable disease severity and cataloged rapidly progressive retinal degeneration. Given the accessibility of neural retina, availability of objective, quantitative readouts for monitoring therapeutic response, and the rapid disease progression in SCA7, ASOs targeting ATAXIN-7 might represent a viable treatment for SCA7 retinal degeneration.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures







References
-
- Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, Szliwowski H, Evrard P, Ceuterick C, Tassignon MJ, Smet-Dieleman H, et al., On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol 88, 277–286 (1994). - PubMed
-
- Filla A, Mariotti C, Caruso G, Coppola G, Cocozza S, Castaldo I, Calabrese O, Salvatore E, De Michele G, Riggio MC, Pareyson D, Gellera C, Di Donato S, Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol 44, 31–36 (2000). - PubMed
-
- Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ, Forrest SM, Biros I, Nicholson GA , Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet 95, 351–357 (2000). - PubMed
-
- Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, et al., Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 378, 403–406. (1995). - PubMed
-
- David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A, Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17, 65–70 (1997). - PubMed
