

Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels
- PMID: 30385745
- PMCID: PMC6212435
- DOI: 10.1038/s41467-018-06891-x
Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels
Erratum in
-
Publisher Correction: Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels.Nat Commun. 2018 Dec 17;9(1):5401. doi: 10.1038/s41467-018-07887-3. Nat Commun. 2018. PMID: 30559342 Free PMC article.
Abstract
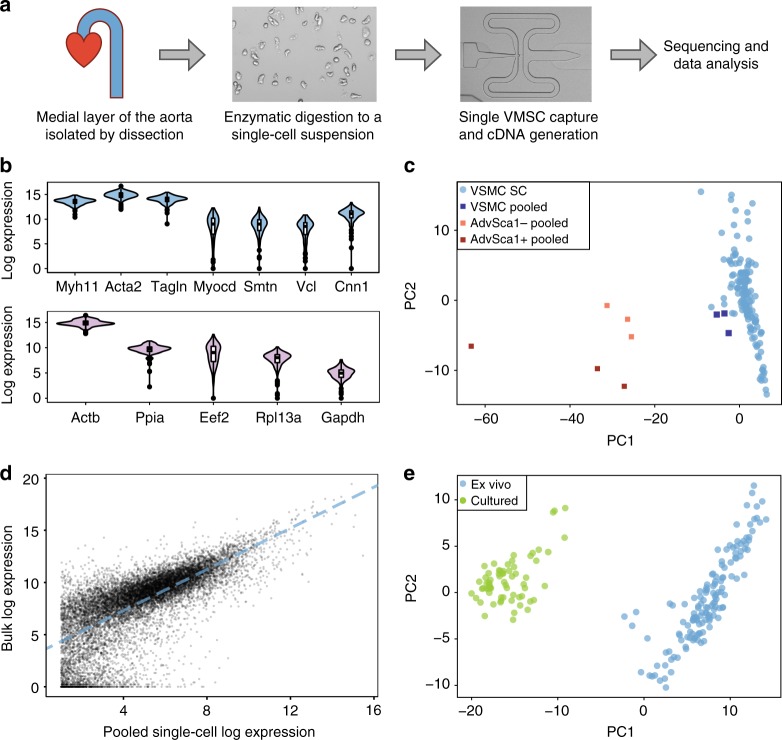
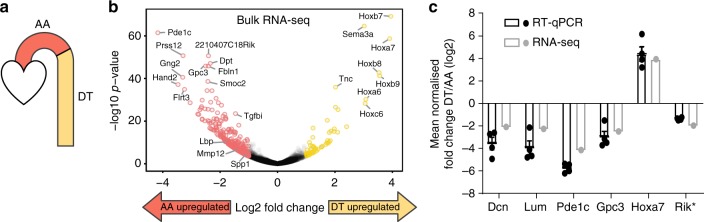
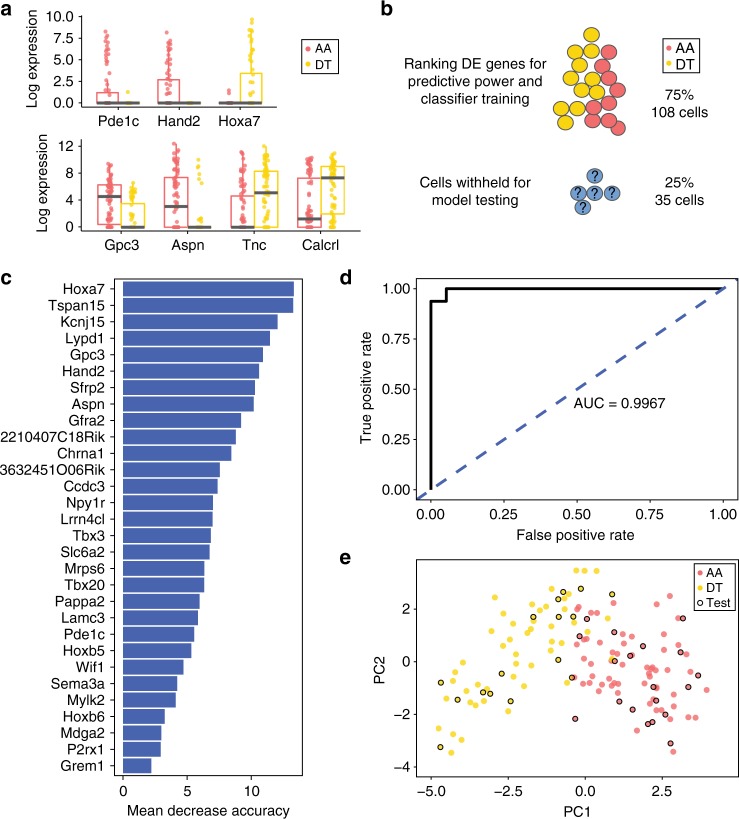
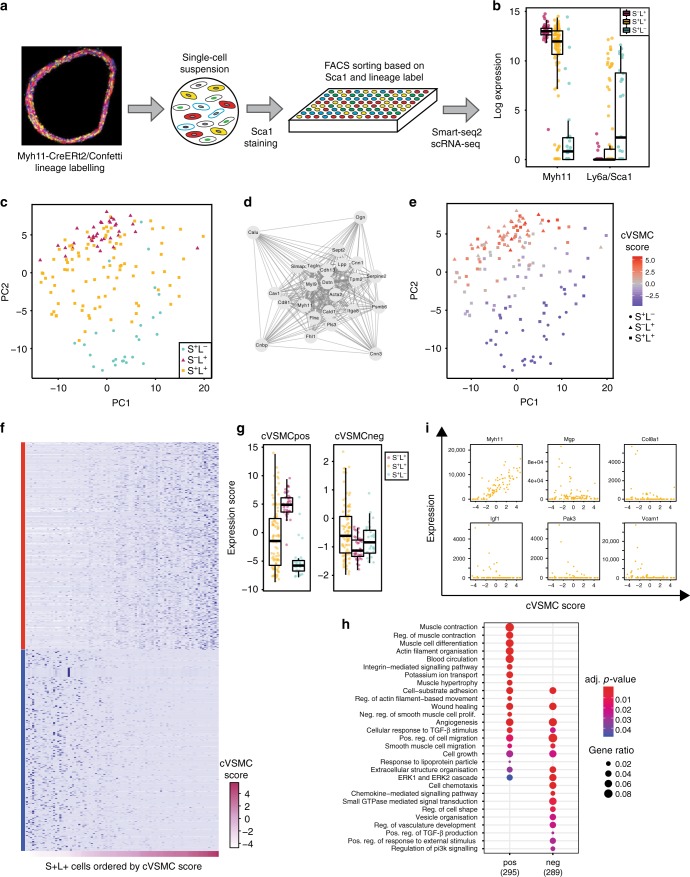
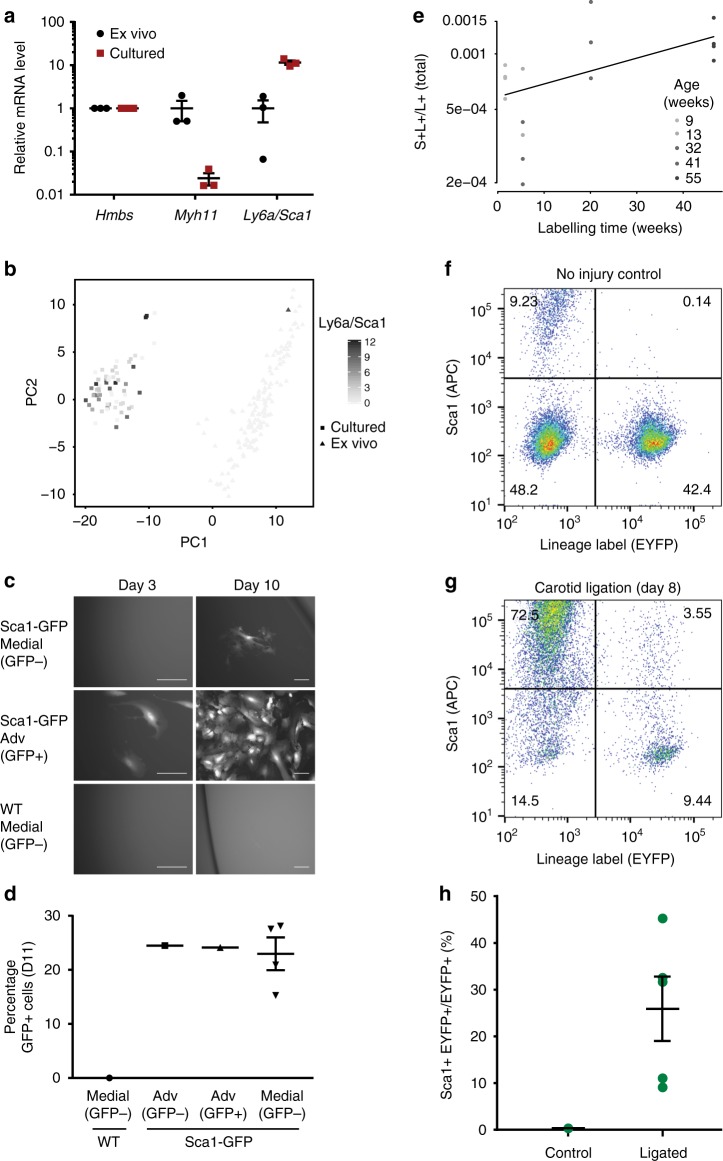
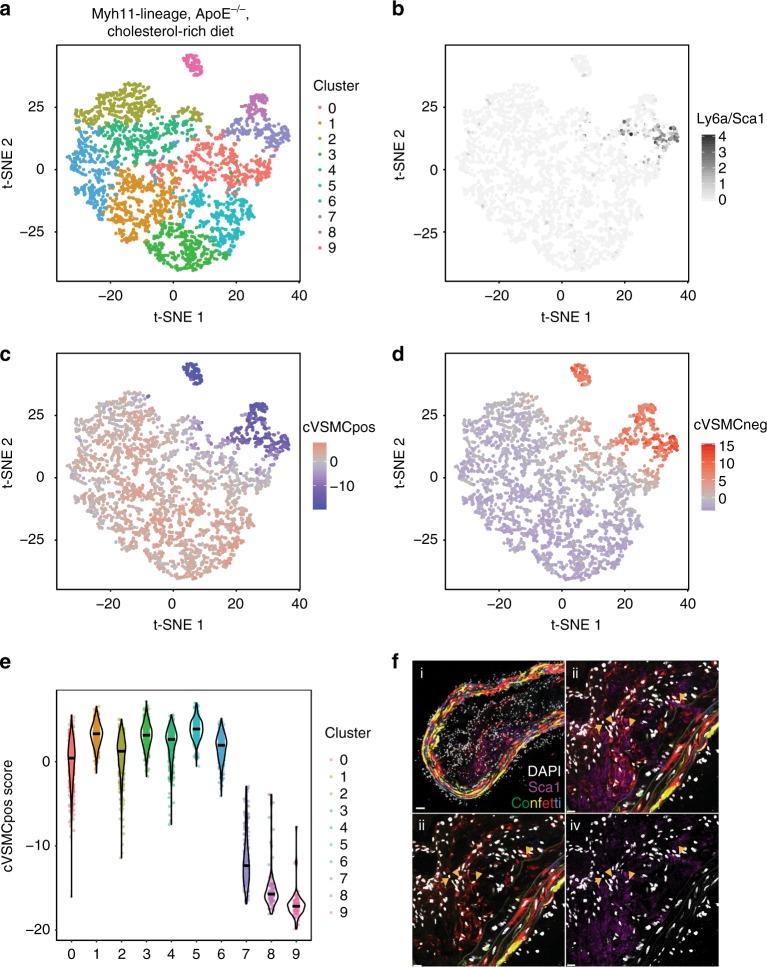
Vascular smooth muscle cells (VSMCs) show pronounced heterogeneity across and within vascular beds, with direct implications for their function in injury response and atherosclerosis. Here we combine single-cell transcriptomics with lineage tracing to examine VSMC heterogeneity in healthy mouse vessels. The transcriptional profiles of single VSMCs consistently reflect their region-specific developmental history and show heterogeneous expression of vascular disease-associated genes involved in inflammation, adhesion and migration. We detect a rare population of VSMC-lineage cells that express the multipotent progenitor marker Sca1, progressively downregulate contractile VSMC genes and upregulate genes associated with VSMC response to inflammation and growth factors. We find that Sca1 upregulation is a hallmark of VSMCs undergoing phenotypic switching in vitro and in vivo, and reveal an equivalent population of Sca1-positive VSMC-lineage cells in atherosclerotic plaques. Together, our analyses identify disease-relevant transcriptional signatures in VSMC-lineage cells in healthy blood vessels, with implications for disease susceptibility, diagnosis and prevention.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Shanahan CM, Weissberg PL. Smooth muscle cell heterogeneity: patterns of gene expression in vascular smooth muscle cells in vitro and in vivo. Cartography. 1998;18:333–338. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- FS/15/38/31516/BHF_/British Heart Foundation/United Kingdom
- RE/13/6/30180/BHF_/British Heart Foundation/United Kingdom
- RM/13/3/30159/BHF_/British Heart Foundation/United Kingdom
- RM/13/3/30159/British Heart Foundation (BHF)/International
- MC_UP_1605/3/MRC_/Medical Research Council/United Kingdom
- FS/14/59/31282/British Heart Foundation (BHF)/International
- PG/16/63/32307/BHF_/British Heart Foundation/United Kingdom
- FS/15/38/31516/British Heart Foundation (BHF)/International
- PG/16/11/32021/BHF_/British Heart Foundation/United Kingdom
- RG/13/14/30314/BHF_/British Heart Foundation/United Kingdom
- PG/12/86/29930/BHF_/British Heart Foundation/United Kingdom
- Core funding/Medical Research Council (MRC)/International
- FS/14/59/31282/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- AdG 341096/EC | European Research Council (ERC)/International
- RE/13/6/30180/British Heart Foundation (BHF)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
