

Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma
- PMID: 30389701
- PMCID: PMC6318036
- DOI: 10.1158/0008-5472.CAN-18-0717
Activation of PP2A and Inhibition of mTOR Synergistically Reduce MYC Signaling and Decrease Tumor Growth in Pancreatic Ductal Adenocarcinoma
Abstract
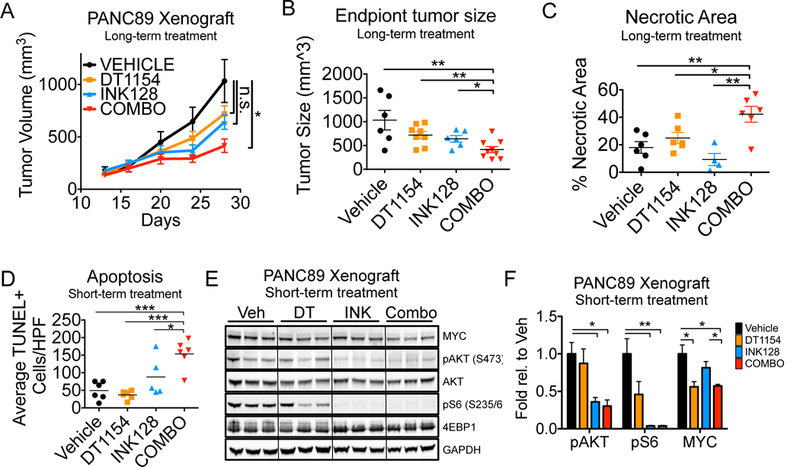
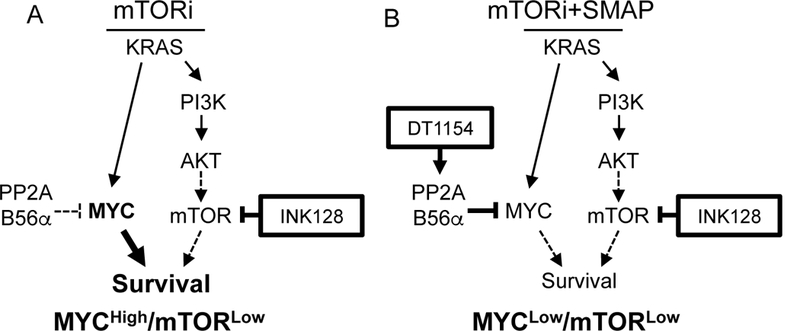
In cancer, kinases are often activated and phosphatases suppressed, leading to aberrant activation of signaling pathways driving cellular proliferation, survival, and therapeutic resistance. Although pancreatic ductal adenocarcinoma (PDA) has historically been refractory to kinase inhibition, therapeutic activation of phosphatases is emerging as a promising strategy to restore balance to these hyperactive signaling cascades. In this study, we hypothesized that phosphatase activation combined with kinase inhibition could deplete oncogenic survival signals to reduce tumor growth. We screened PDA cell lines for kinase inhibitors that could synergize with activation of protein phosphatase 2A (PP2A), a tumor suppressor phosphatase, and determined that activation of PP2A and inhibition of mTOR synergistically increase apoptosis and reduce oncogenic phenotypes in vitro and in vivo. This combination treatment resulted in suppression of AKT/mTOR signaling coupled with reduced expression of c-MYC, an oncoprotein implicated in tumor progression and therapeutic resistance. Forced expression of c-MYC or loss of PP2A B56α, the specific PP2A subunit shown to negatively regulate c-MYC, increased resistance to mTOR inhibition. Conversely, decreased c-MYC expression increased the sensitivity of PDA cells to mTOR inhibition. Together, these studies demonstrate that combined targeting of PP2A and mTOR suppresses proliferative signaling and induces cell death and implicates this combination as a promising therapeutic strategy for patients with PDA. SIGNIFICANCE: These findings present a combinatorial strategy targeting serine/threonine protein phosphatase PP2A and mTOR in PDA, a cancer for which there are currently no targeted therapeutic options.Graphical Abstract: http://cancerres.aacrjournals.org/content/canres/79/1/209/F1.large.jpg.
©2018 American Association for Cancer Research.
Conflict of interest statement
Figures
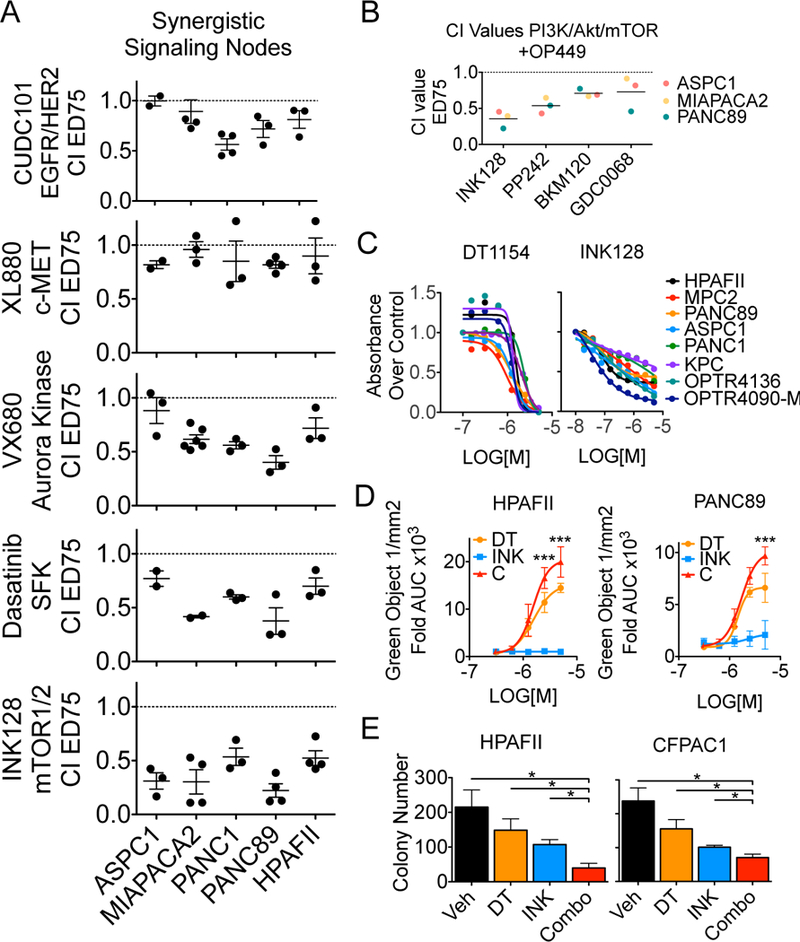
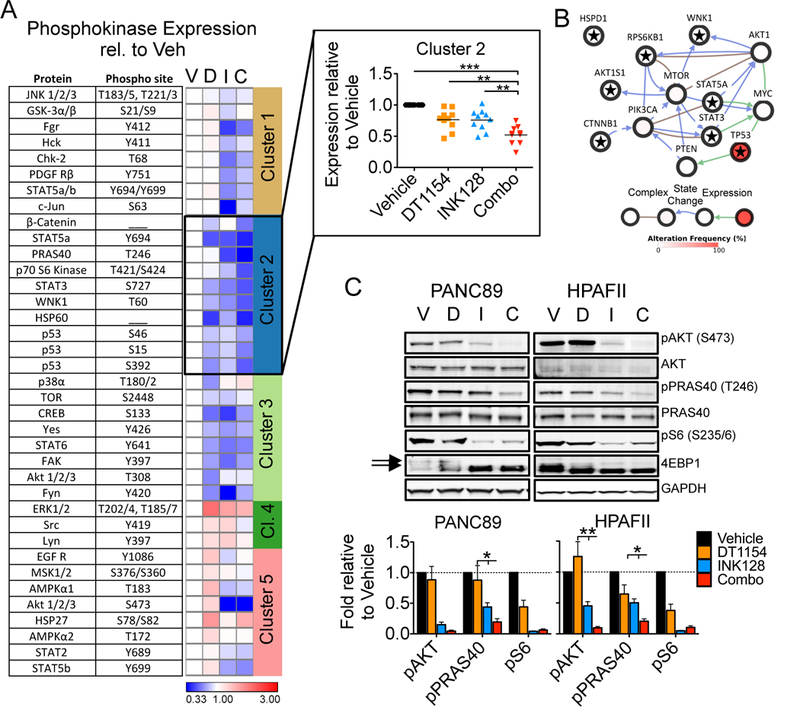
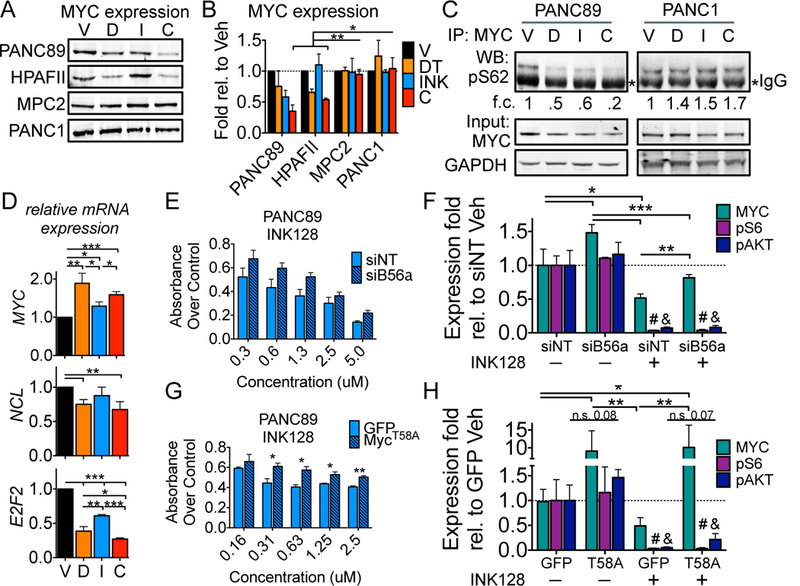
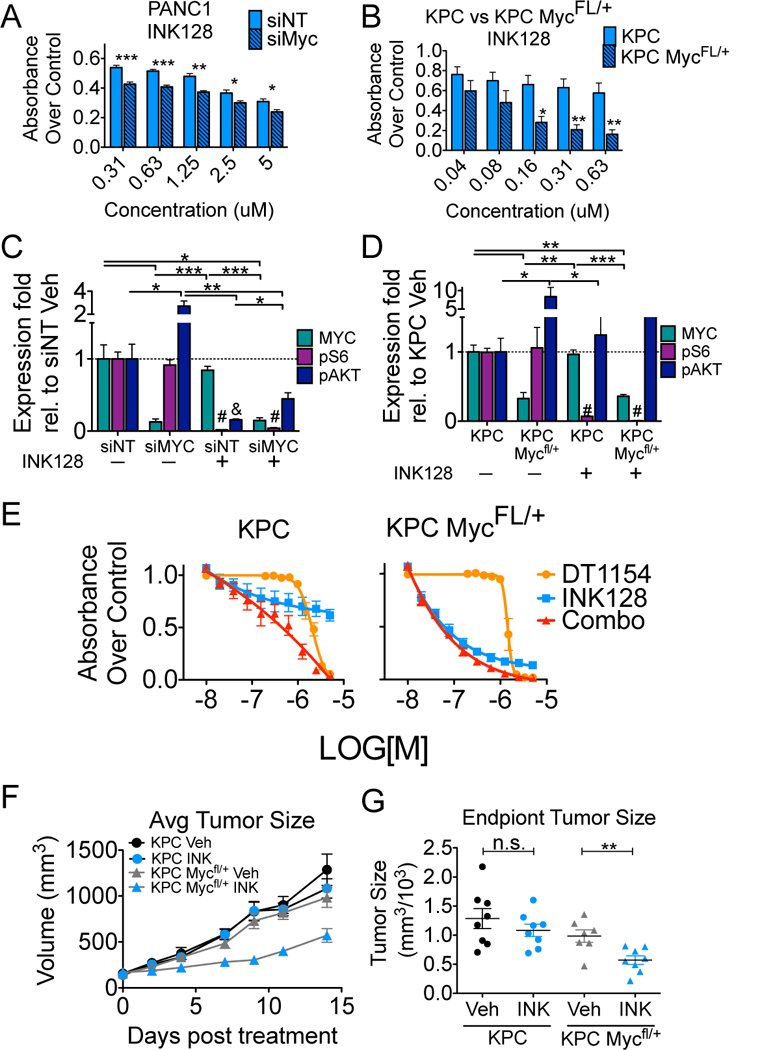
References
-
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
