

Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement
- PMID: 30389915
- PMCID: PMC6214957
- DOI: 10.1038/s41467-018-06983-8
Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement
Abstract
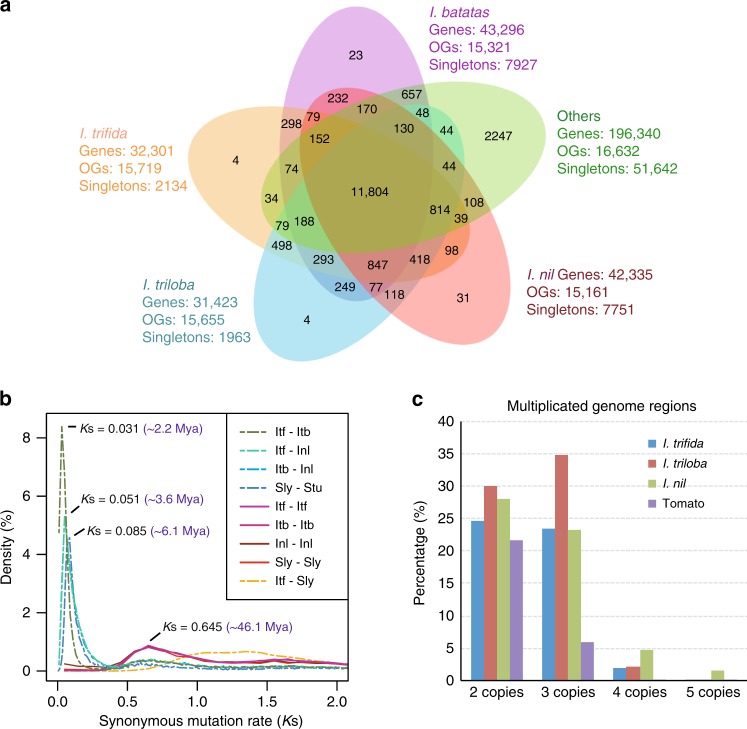
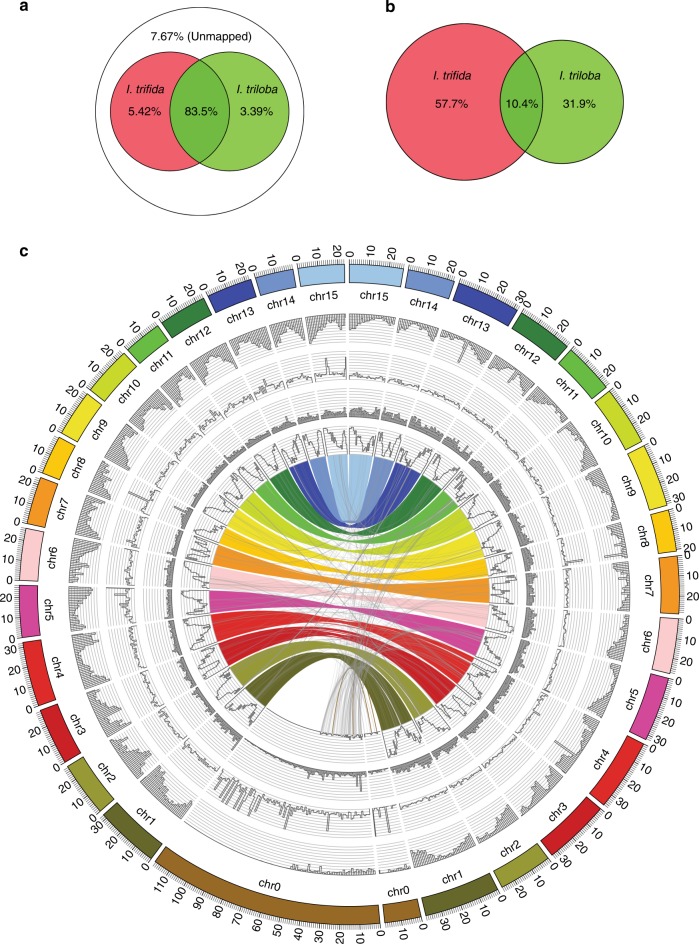
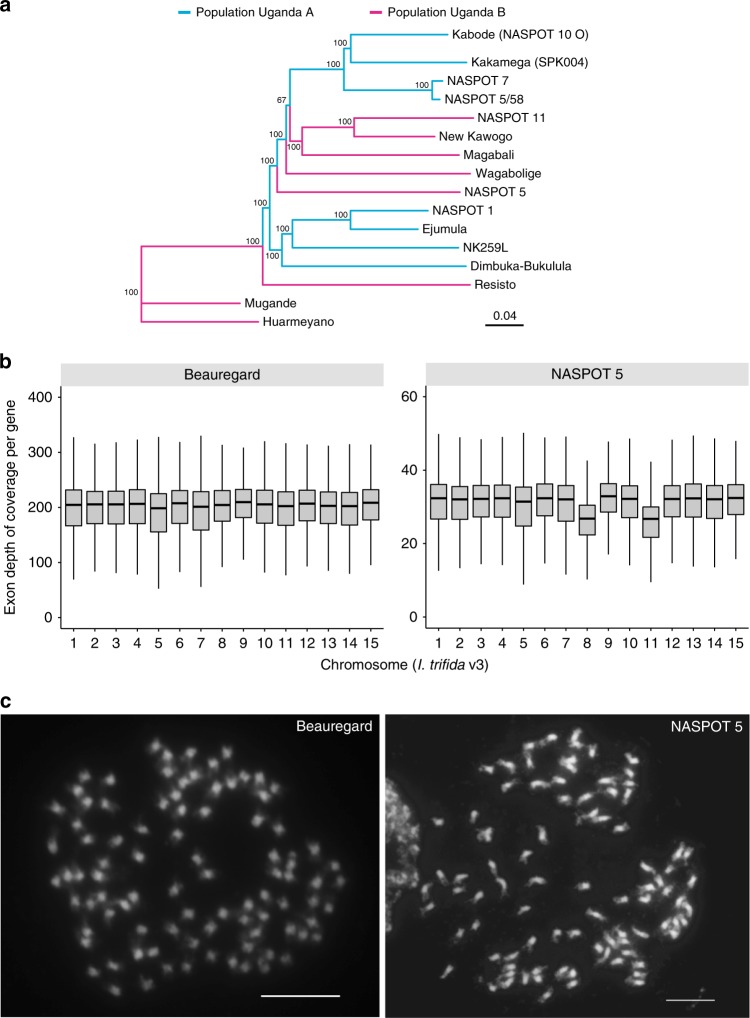
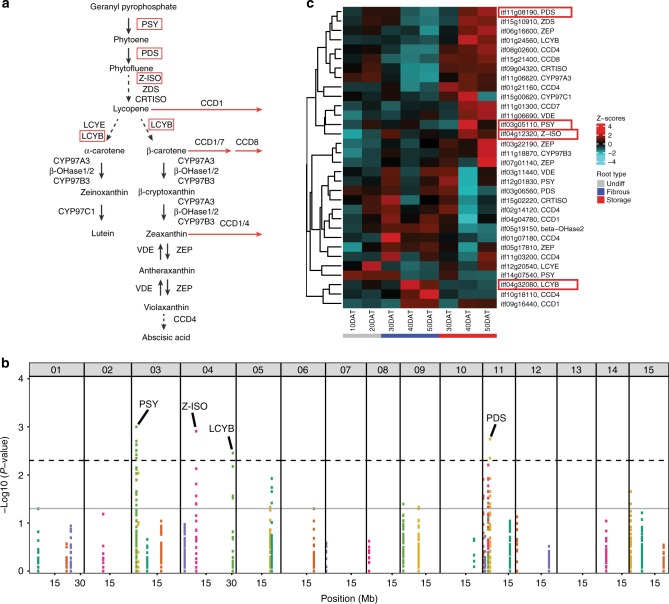
Sweetpotato [Ipomoea batatas (L.) Lam.] is a globally important staple food crop, especially for sub-Saharan Africa. Agronomic improvement of sweetpotato has lagged behind other major food crops due to a lack of genomic and genetic resources and inherent challenges in breeding a heterozygous, clonally propagated polyploid. Here, we report the genome sequences of its two diploid relatives, I. trifida and I. triloba, and show that these high-quality genome assemblies are robust references for hexaploid sweetpotato. Comparative and phylogenetic analyses reveal insights into the ancient whole-genome triplication history of Ipomoea and evolutionary relationships within the Batatas complex. Using resequencing data from 16 genotypes widely used in African breeding programs, genes and alleles associated with carotenoid biosynthesis in storage roots are identified, which may enable efficient breeding of varieties with high provitamin A content. These resources will facilitate genome-enabled breeding in this important food security crop.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Loebenstein, G. in The Sweetpotato (eds Loebenstein, G. & Thottappilly, G. eds.) 9–12 (Springer, 2009).
-
- Labarta, R. in Crop Improvement, Adoption, and Impact of Improved Varieties in Food Crops in Sub-Saharan Africa (eds. Walker, T. S. & Alwang, J.) 164–182 (CABI, 2015).
-
- Austin, D. F. in Exploration, Maintenance and Utilization of Sweet Potato Genetic Resources. Report of the First Sweet Potato Planning Conference (International Potato Centre (CIP), Lima, Peru, 1987) (ed. P., G.) 27–59 (CIP, 1988).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
