

Hematopoietic Stem Cell Transplantation as Treatment for Patients with DOCK8 Deficiency
- PMID: 30391550
- PMCID: PMC6771433
- DOI: 10.1016/j.jaip.2018.10.035
Hematopoietic Stem Cell Transplantation as Treatment for Patients with DOCK8 Deficiency
Abstract
Background: Biallelic variations in the dedicator of cytokinesis 8 (DOCK8) gene cause a combined immunodeficiency with eczema, recurrent bacterial and viral infections, and malignancy. Natural disease outcome is dismal, but allogeneic hematopoietic stem cell transplantation (HSCT) can cure the disease.
Objective: To determine outcome of HSCT for DOCK8 deficiency and define possible outcome variables.
Methods: We performed a retrospective study of the results of HSCT in a large international cohort of DOCK8-deficient patients.
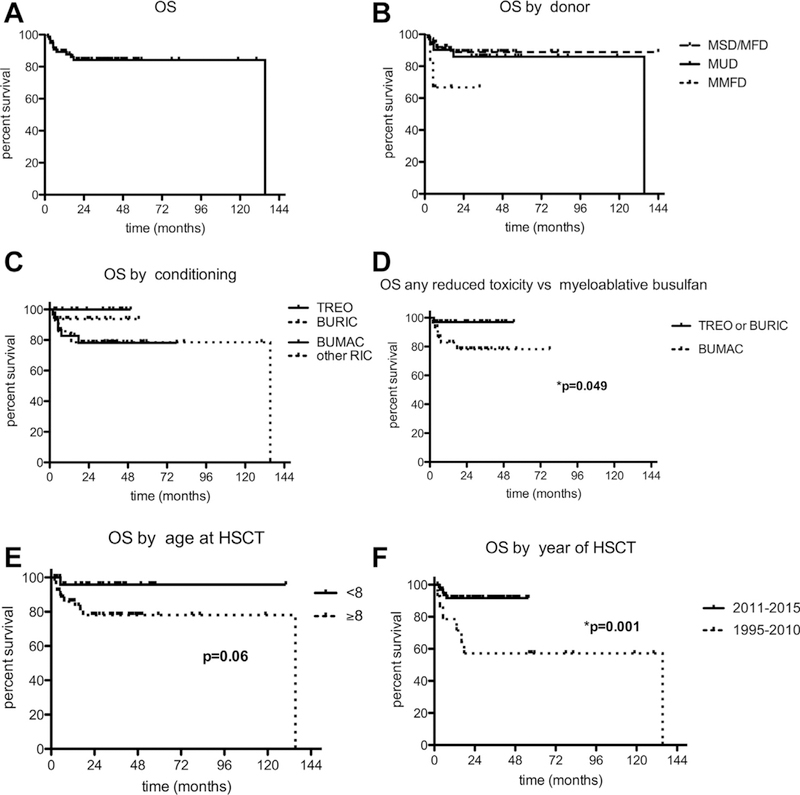
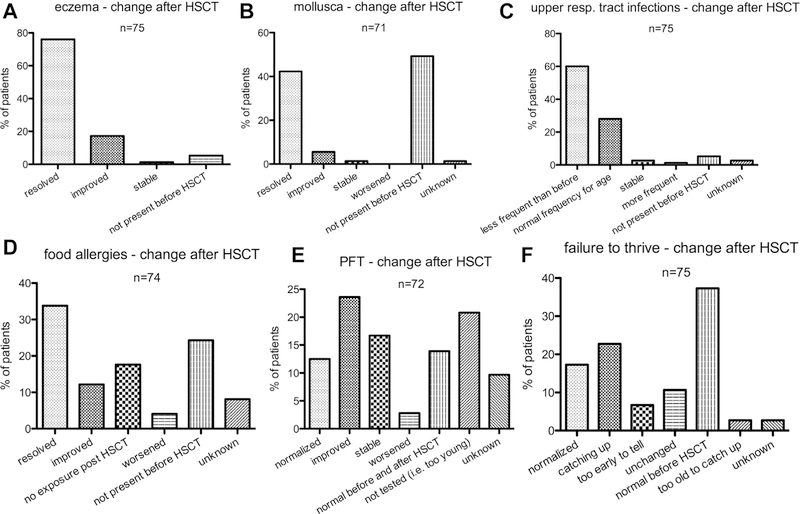
Results: We identified 81 patients from 22 centers transplanted at a median age of 9.7 years (range, 0.7-27.2 years) between 1995 and 2015. After median follow-up of 26 months (range, 3-135 months), 68 (84%) patients are alive. Severe acute (III-IV) or chronic graft versus host disease occurred in 11% and 10%, respectively. Causes of death were infections (n = 5), graft versus host disease (5), multiorgan failure (2), and preexistent lymphoma (1). Survival after matched related (n = 40) or unrelated (35) HSCT was 89% and 81%, respectively. Reduced-toxicity conditioning based on either treosulfan or reduced-dose busulfan resulted in superior survival compared with fully myeloablative busulfan-based regimens (97% vs 78%; P = .049). Ninety-six percent of patients younger than 8 years at HSCT survived, compared with 78% of those 8 years and older (P = .06). Of the 73 patients with chimerism data available, 65 (89%) had more than 90% donor T-cell chimerism at last follow-up. Not all disease manifestations responded equally well to HSCT: eczema, infections, and mollusca resolved quicker than food allergies or failure to thrive.
Conclusions: HSCT is curative in most DOCK8-deficient patients, confirming this approach as the treatment of choice. HSCT using a reduced-toxicity regimen may offer the best chance for survival.
Keywords: Combined immunodeficiency; DOCK8 deficiency; HSCT.
Copyright © 2018. Published by Elsevier Inc.
Conflict of interest statement
Conflicts of interest: The rest of the authors declare that they have no relevant conflicts of interest.
Figures

References
-
- Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr 2004;144:93–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
