

Bayesian Weighing of Electron Cryo-Microscopy Data for Integrative Structural Modeling
- PMID: 30393052
- PMCID: PMC6779587
- DOI: 10.1016/j.str.2018.09.011
Bayesian Weighing of Electron Cryo-Microscopy Data for Integrative Structural Modeling
Abstract
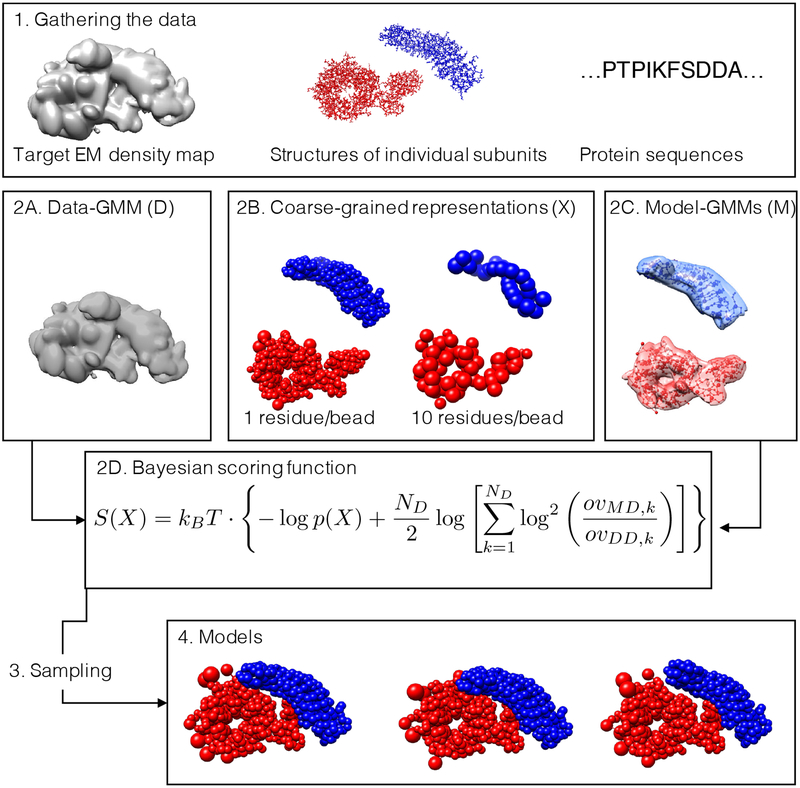
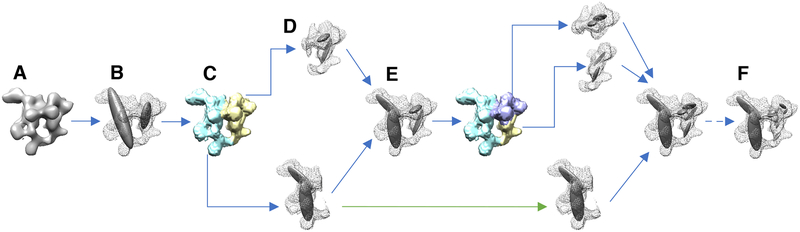
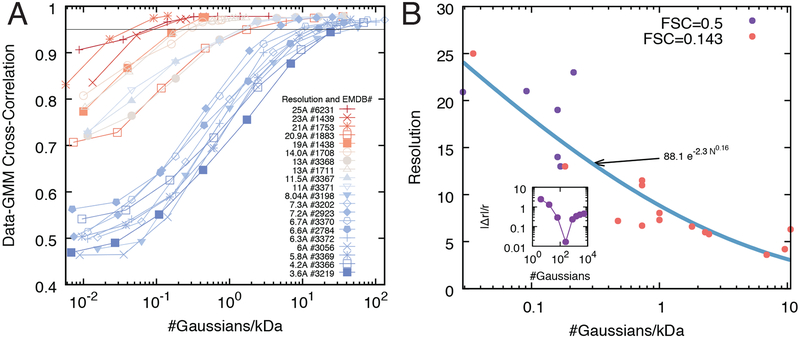
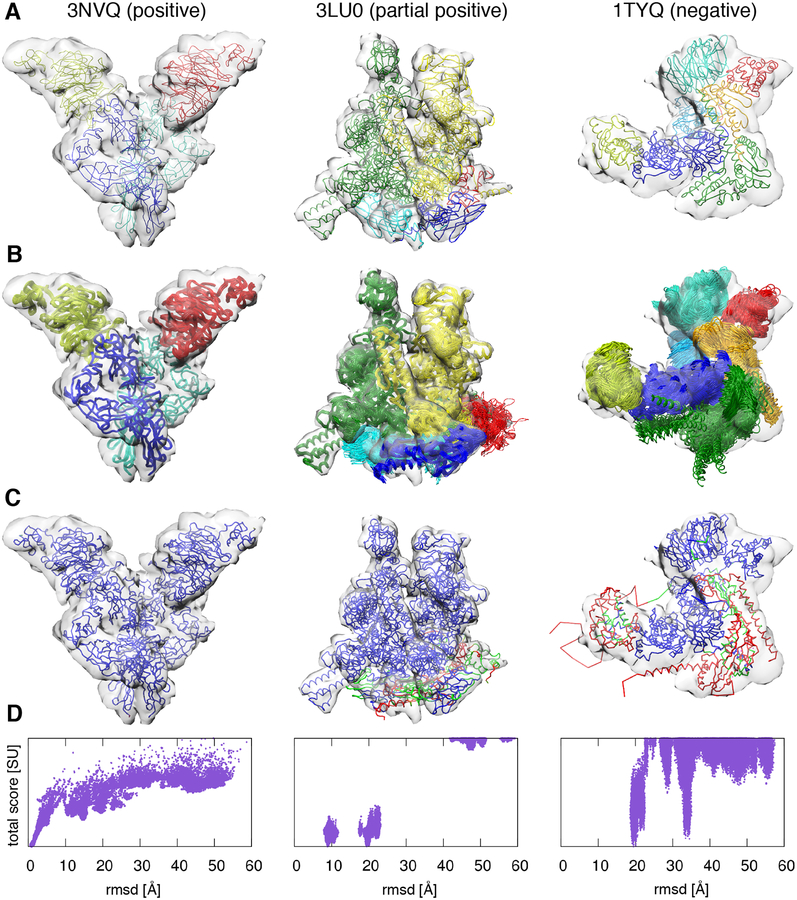
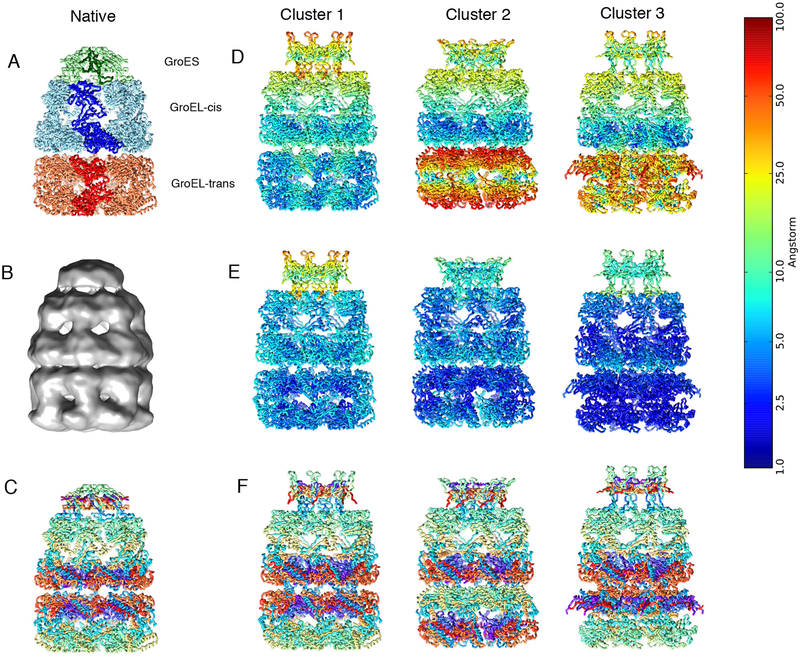
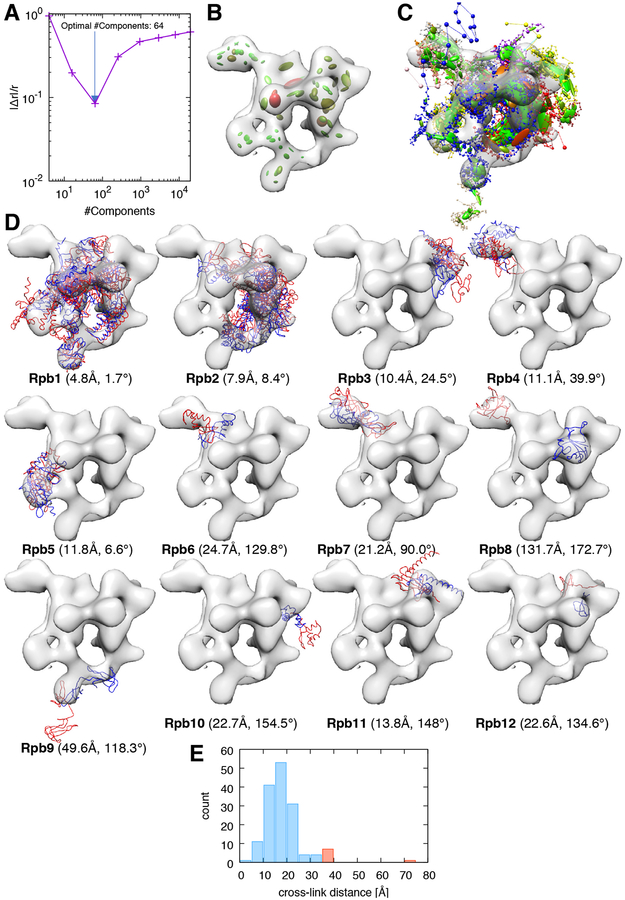
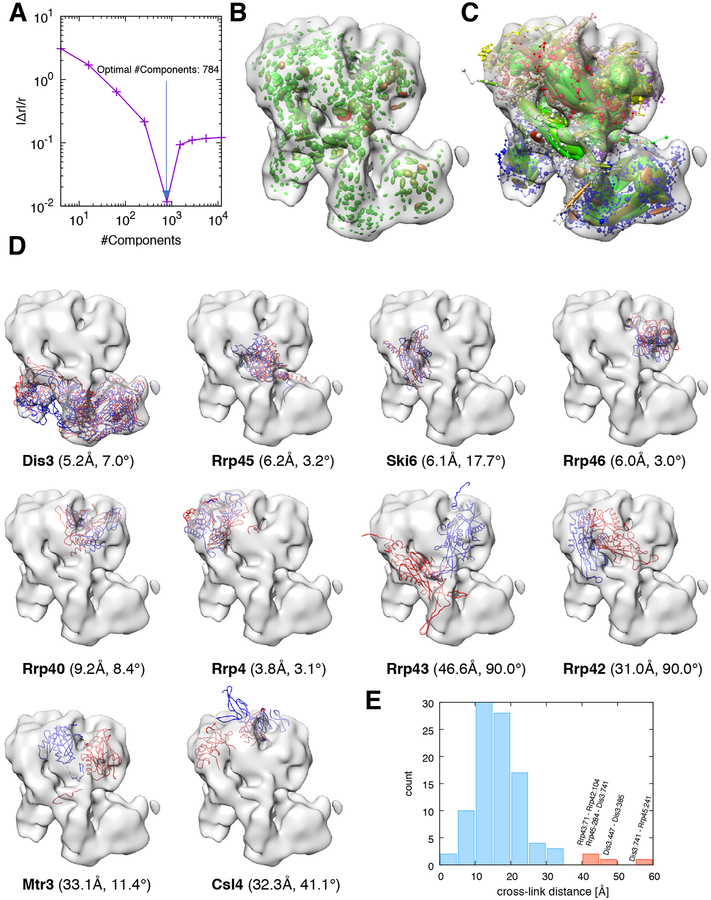
Cryo-electron microscopy (cryo-EM) has become a mainstream technique for determining the structures of complex biological systems. However, accurate integrative structural modeling has been hampered by the challenges in objectively weighing cryo-EM data against other sources of information due to the presence of random and systematic errors, as well as correlations, in the data. To address these challenges, we introduce a Bayesian scoring function that efficiently and accurately ranks alternative structural models of a macromolecular system based on their consistency with a cryo-EM density map as well as other experimental and prior information. The accuracy of this approach is benchmarked using complexes of known structure and illustrated in three applications: the structural determination of the GroEL/GroES, RNA polymerase II, and exosome complexes. The approach is implemented in the open-source Integrative Modeling Platform (http://integrativemodeling.org), thus enabling integrative structure determination by combining cryo-EM data with other sources of information.
Keywords: Gaussian mixture model; bayesian inference; cross-linking mass spectrometry; cryo-electron microscopy; data weighing; integrative structural modeling; macromolecular complexes; structural biology.
Copyright © 2018. Published by Elsevier Ltd.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures
References
-
- Armache K-J, Mitterweger S, Meinhart A, and Cramer P (2005). Structures of Complete RNA Polymerase II and its Subcomplex, Rpb4/7. J Biol Chem 280, 7131. - PubMed
-
- Bai XC, McMullan G, and Scheres SH (2015). How cryo-EM is revolutionizing structural biology. Trends in Biochemical Sciences 40, 49–57. - PubMed
-
- Bernecky C, Herzog F, Baumeister W, Plitzko JM, and Cramer P (2016). Structure of transcribing mammalian RNA polymerase II. Nature 529, 551-+. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
