

Widespread Selection for Oncogenic Mutant Allele Imbalance in Cancer
- PMID: 30393068
- PMCID: PMC6234065
- DOI: 10.1016/j.ccell.2018.10.003
Widespread Selection for Oncogenic Mutant Allele Imbalance in Cancer
Abstract
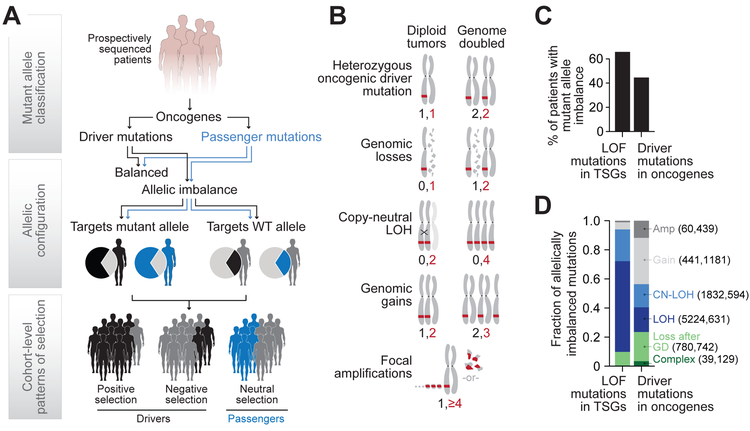
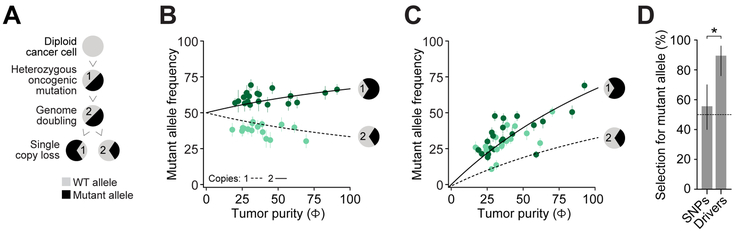
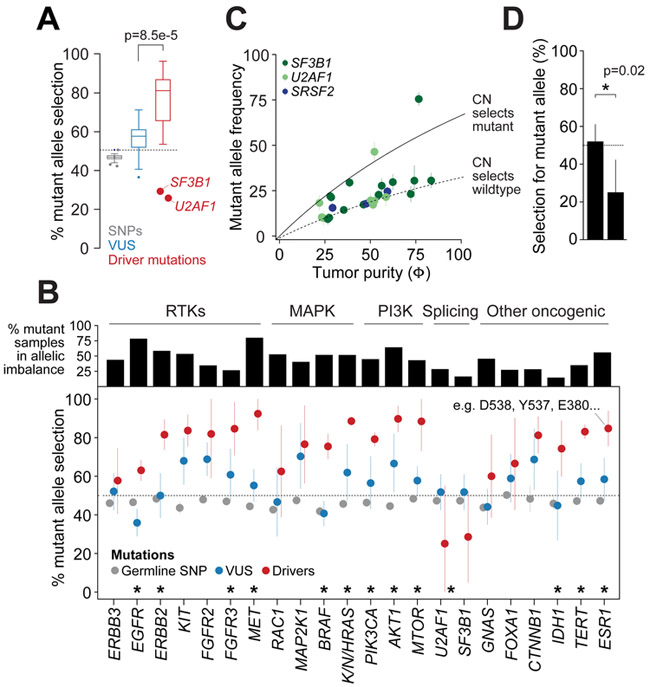
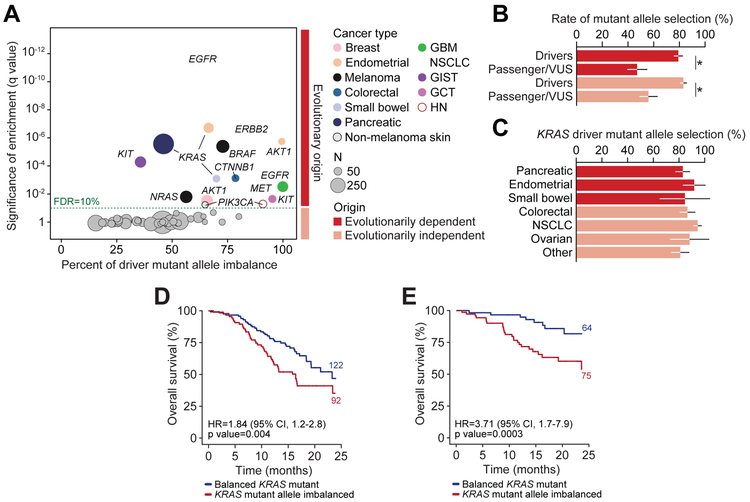
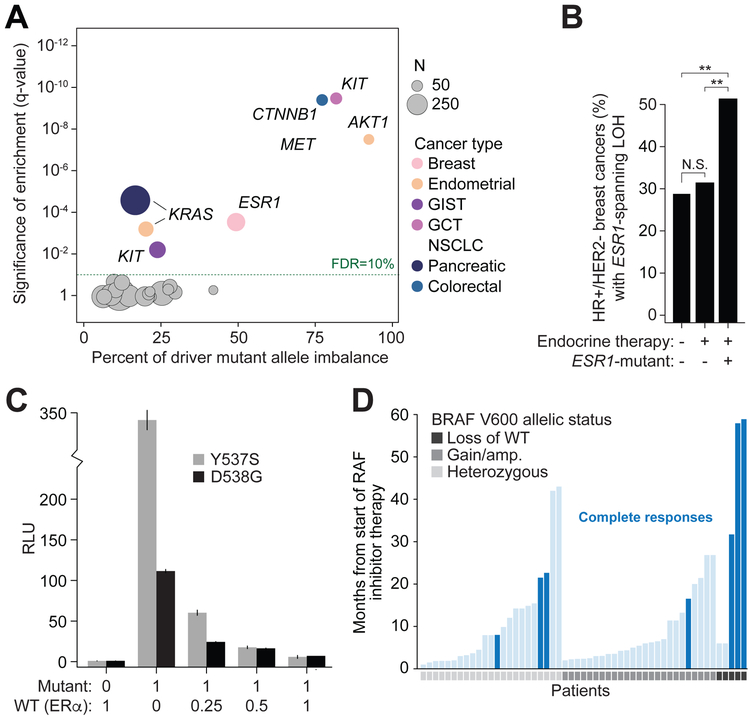
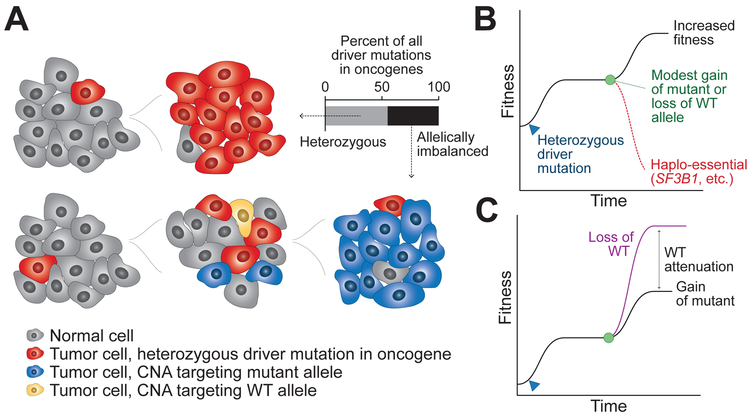
Driver mutations in oncogenes encode proteins with gain-of-function properties that enhance fitness. Heterozygous mutations are thus viewed as sufficient for tumorigenesis. We describe widespread oncogenic mutant allele imbalance in 13,448 prospectively characterized cancers. Imbalance was selected for through modest dosage increases of gain-of-fitness mutations. Negative selection targeted haplo-essential effectors of the spliceosome. Loss of the normal allele comprised a distinct class of imbalance driven by competitive fitness, which correlated with enhanced response to targeted therapies. In many cancers, an antecedent oncogenic mutation drove evolutionarily dependent allele-specific imbalance. In other instances, oncogenic mutations co-opted independent copy-number changes via the evolutionary process of exaptation. Oncogenic allele imbalance is a pervasive evolutionary innovation that enhances fitness and modulates sensitivity to targeted therapy.
Keywords: cancer; competitive fitness; exaptation; oncogenes; selection; targeted therapy.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Ambrogio C, Kohler J, Zhou ZW, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al. (2018). KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 172, 857–868 e815. - PubMed
-
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57, 289–300.
-
- Bentley C, Jurinka SS, Kljavin NM, Vartanian S, Ramani SR, Gonzalez LC, Yu K, Modrusan Z, Du P, Bourgon R, et al. (2013). A requirement for wild-type Ras isoforms in mutant KRas-driven signalling and transformation. Biochem J 452, 313–320. - PubMed
-
- Bremner R, and Balmain A (1990). Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell 61, 407–417. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
