

i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks
- PMID: 30393778
- PMCID: PMC6208412
- DOI: 10.1038/s42003-018-0165-9
i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks
Abstract
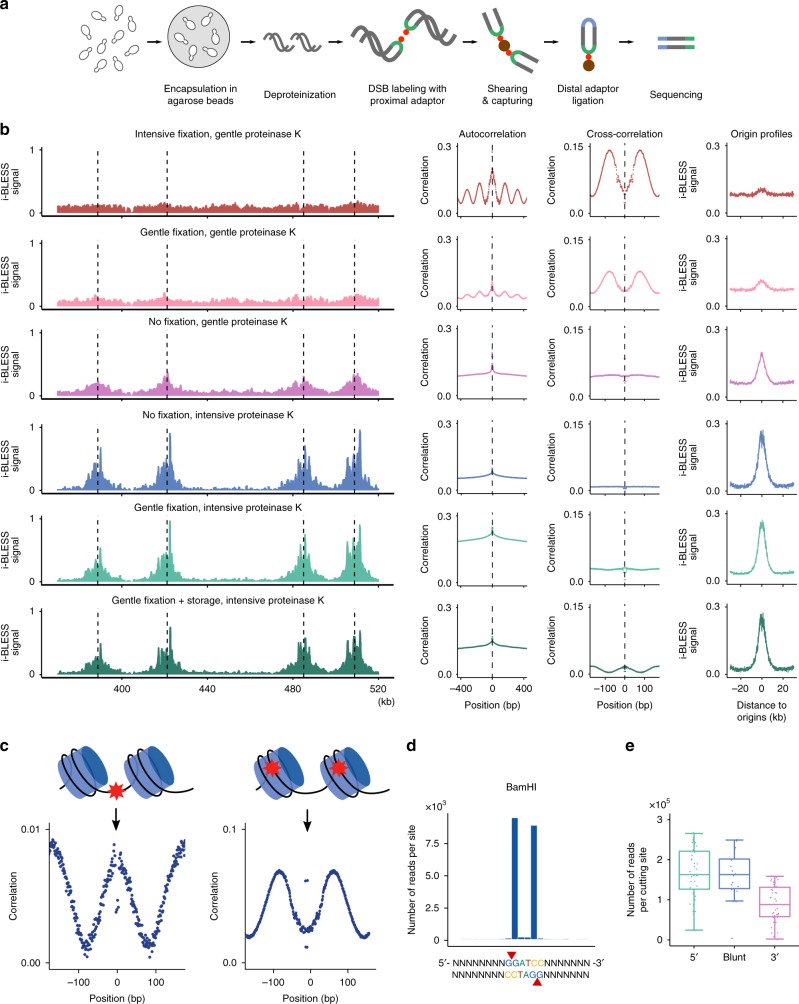
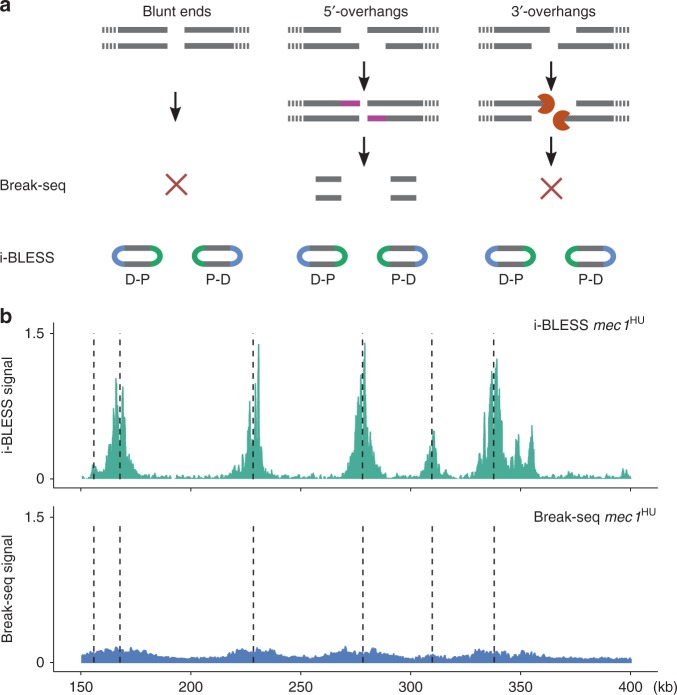
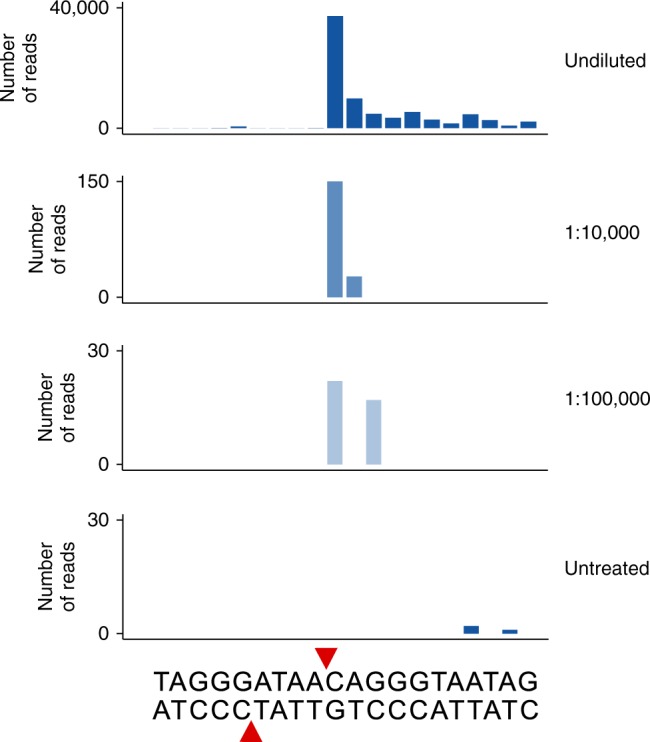
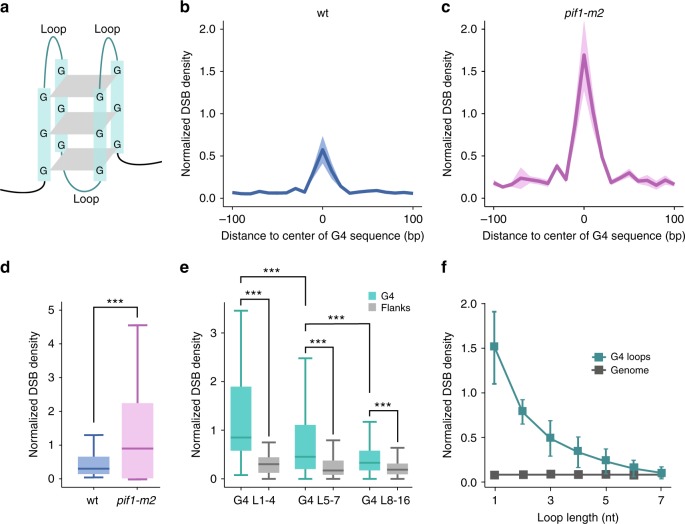
Maintenance of genome stability is a key issue for cell fate that could be compromised by chromosome deletions and translocations caused by DNA double-strand breaks (DSBs). Thus development of precise and sensitive tools for DSBs labeling is of great importance for understanding mechanisms of DSB formation, their sensing and repair. Until now there has been no high resolution and specific DSB detection technique that would be applicable to any cells regardless of their size. Here, we present i-BLESS, a universal method for direct genome-wide DNA double-strand break labeling in cells immobilized in agarose beads. i-BLESS has three key advantages: it is the only unbiased method applicable to yeast, achieves a sensitivity of one break at a given position in 100,000 cells, and eliminates background noise while still allowing for fixation of samples. The method allows detection of ultra-rare breaks such as those forming spontaneously at G-quadruplexes.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
