

Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences
- PMID: 30395178
- PMCID: PMC6581436
- DOI: 10.1093/bioinformatics/bty895
Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences
Abstract
Motivation: In RNA-seq differential expression analysis, investigators aim to detect those genes with changes in expression level across conditions, despite technical and biological variability in the observations. A common task is to accurately estimate the effect size, often in terms of a logarithmic fold change (LFC).
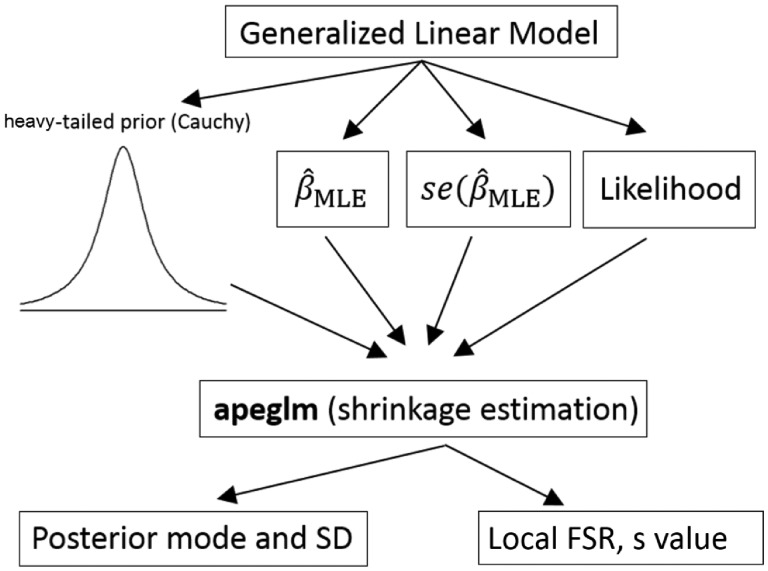
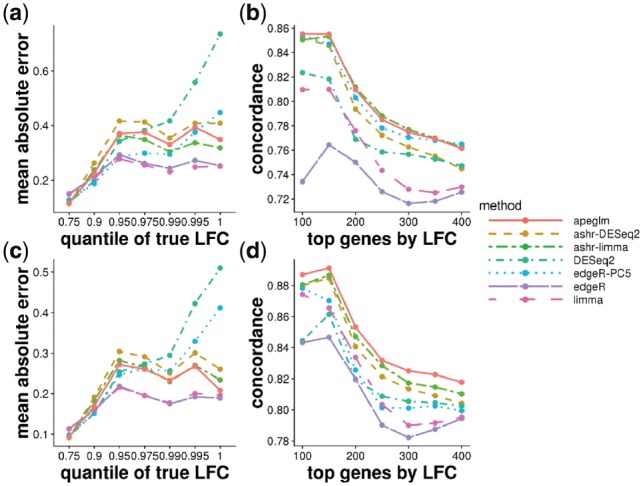
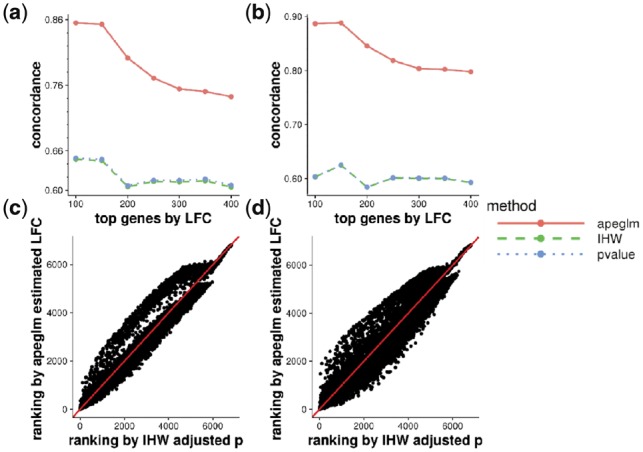
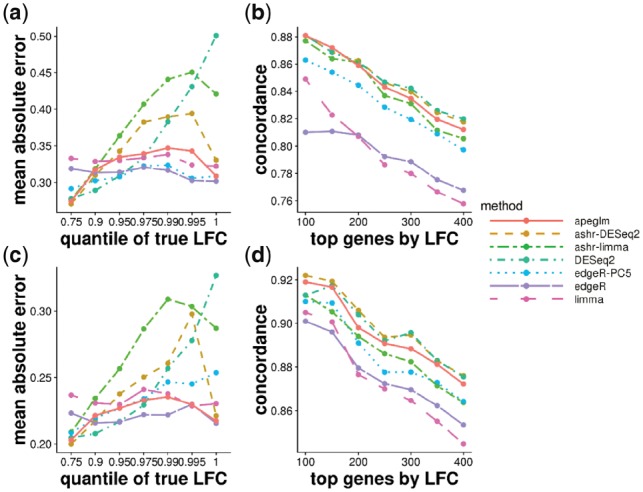
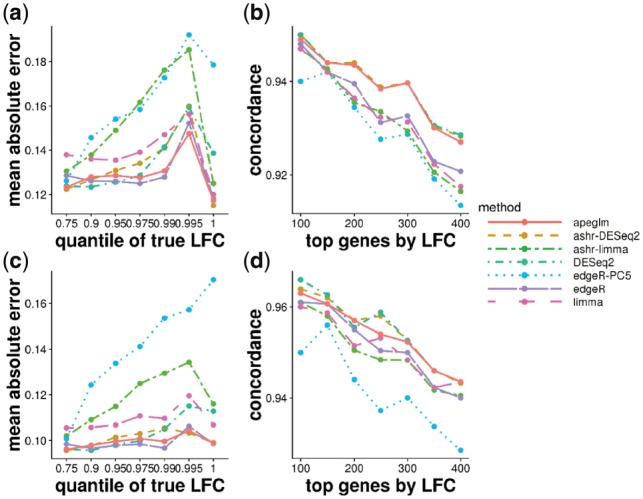
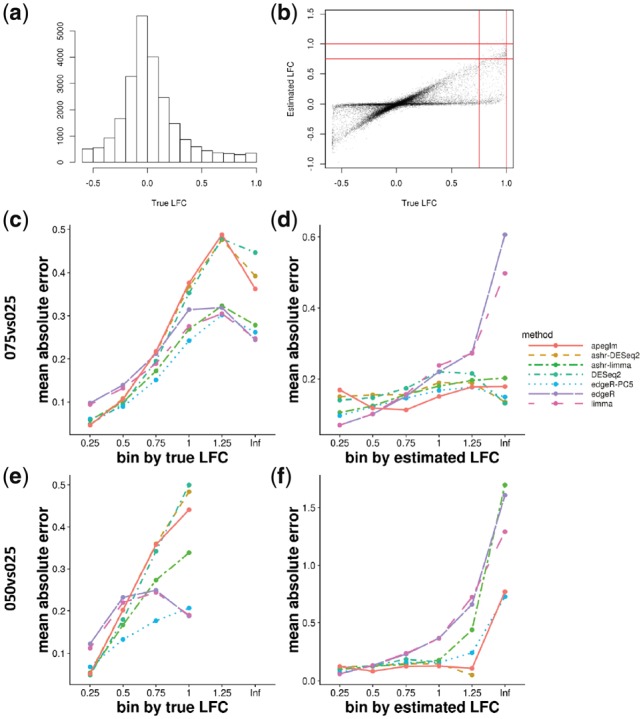
Results: When the read counts are low or highly variable, the maximum likelihood estimates for the LFCs has high variance, leading to large estimates not representative of true differences, and poor ranking of genes by effect size. One approach is to introduce filtering thresholds and pseudocounts to exclude or moderate estimated LFCs. Filtering may result in a loss of genes from the analysis with true differences in expression, while pseudocounts provide a limited solution that must be adapted per dataset. Here, we propose the use of a heavy-tailed Cauchy prior distribution for effect sizes, which avoids the use of filter thresholds or pseudocounts. The proposed method, Approximate Posterior Estimation for generalized linear model, apeglm, has lower bias than previously proposed shrinkage estimators, while still reducing variance for those genes with little information for statistical inference.
Availability and implementation: The apeglm package is available as an R/Bioconductor package at https://bioconductor.org/packages/apeglm, and the methods can be called from within the DESeq2 software.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2018. Published by Oxford University Press.
Figures
References
-
- Brent R.P. (1972). Algorithms for Minimization without Derivatives. Prentice-Hall, Englewood Cliffs, New Jersey, 1973.
-
- Choi H. et al. (2008) Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res., 7, 286–292. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
