

An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion
- PMID: 30395540
- PMCID: PMC6264631
- DOI: 10.1172/JCI123360
An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion
Abstract
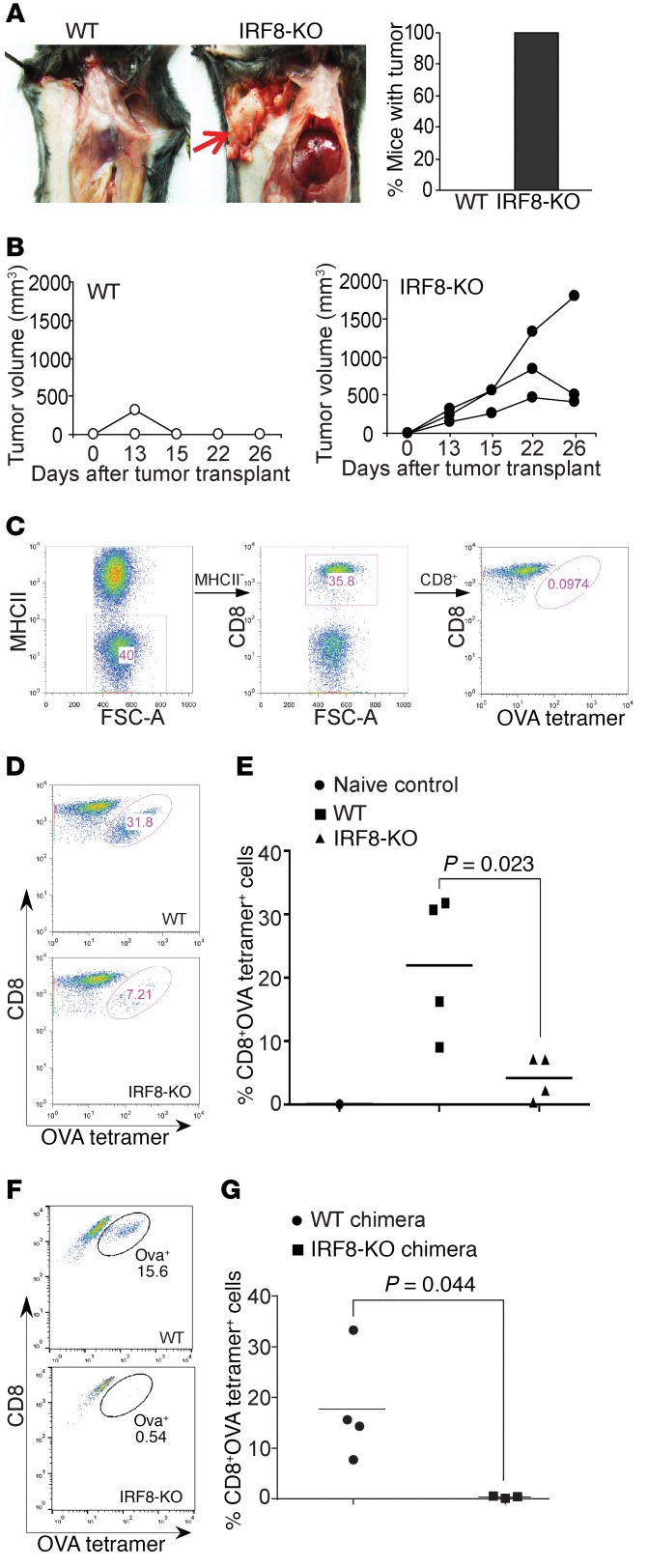
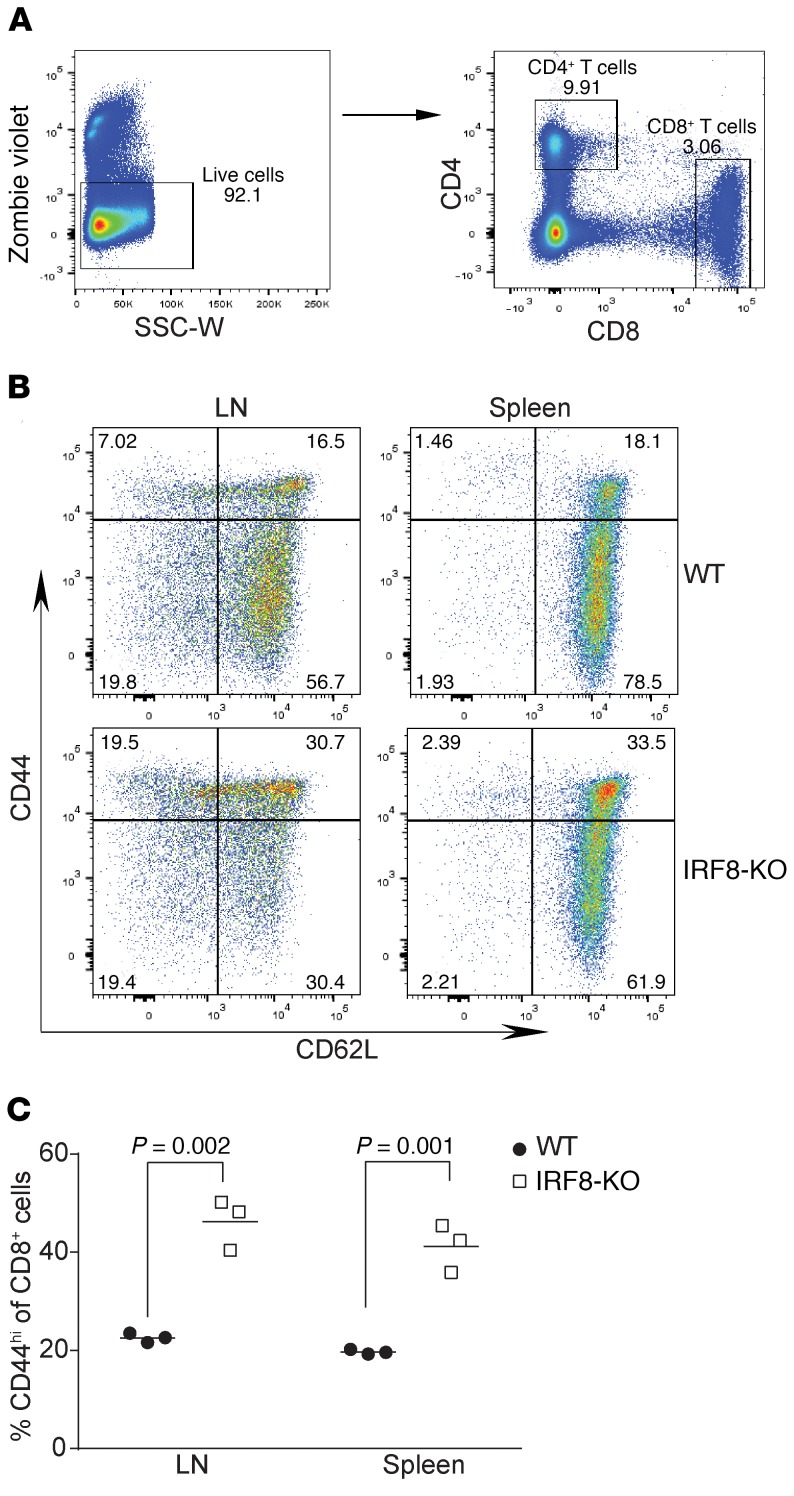
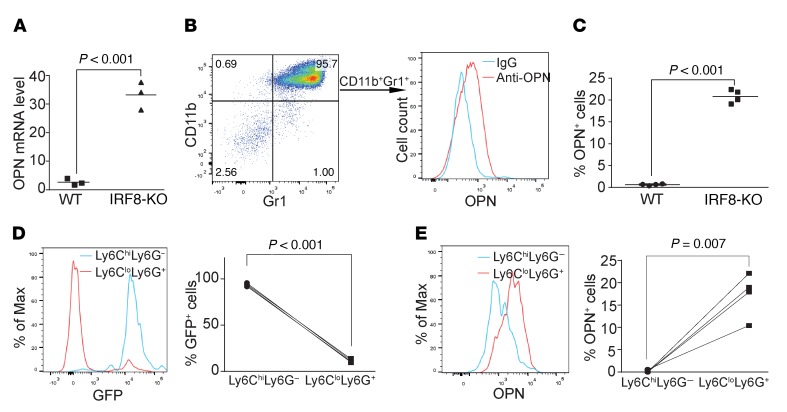
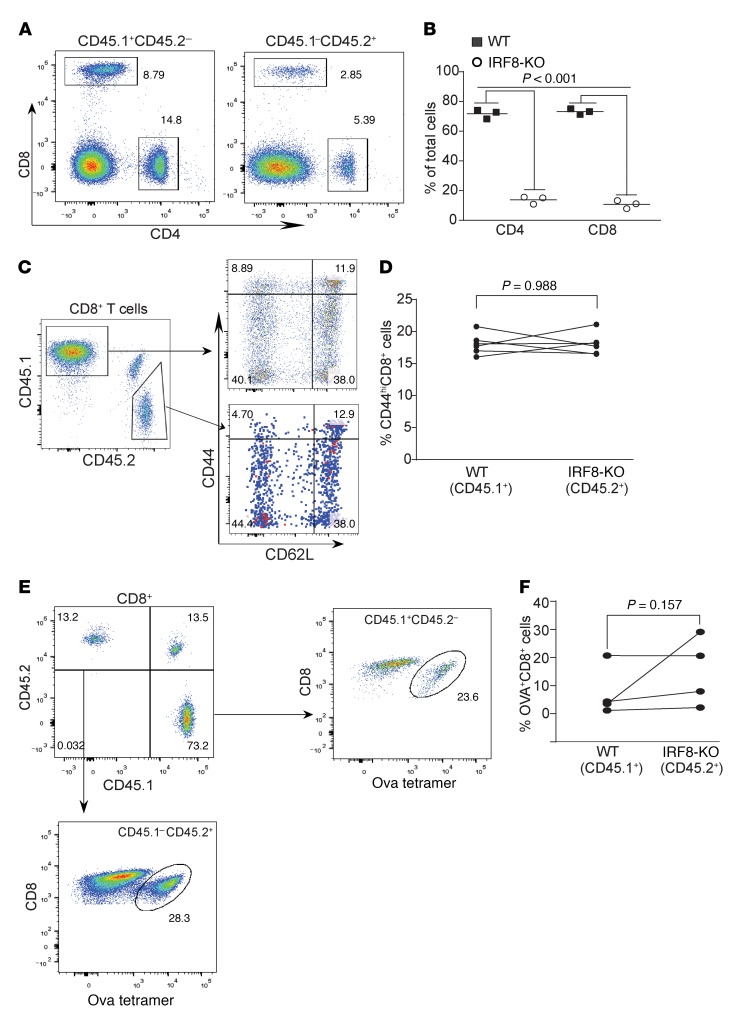
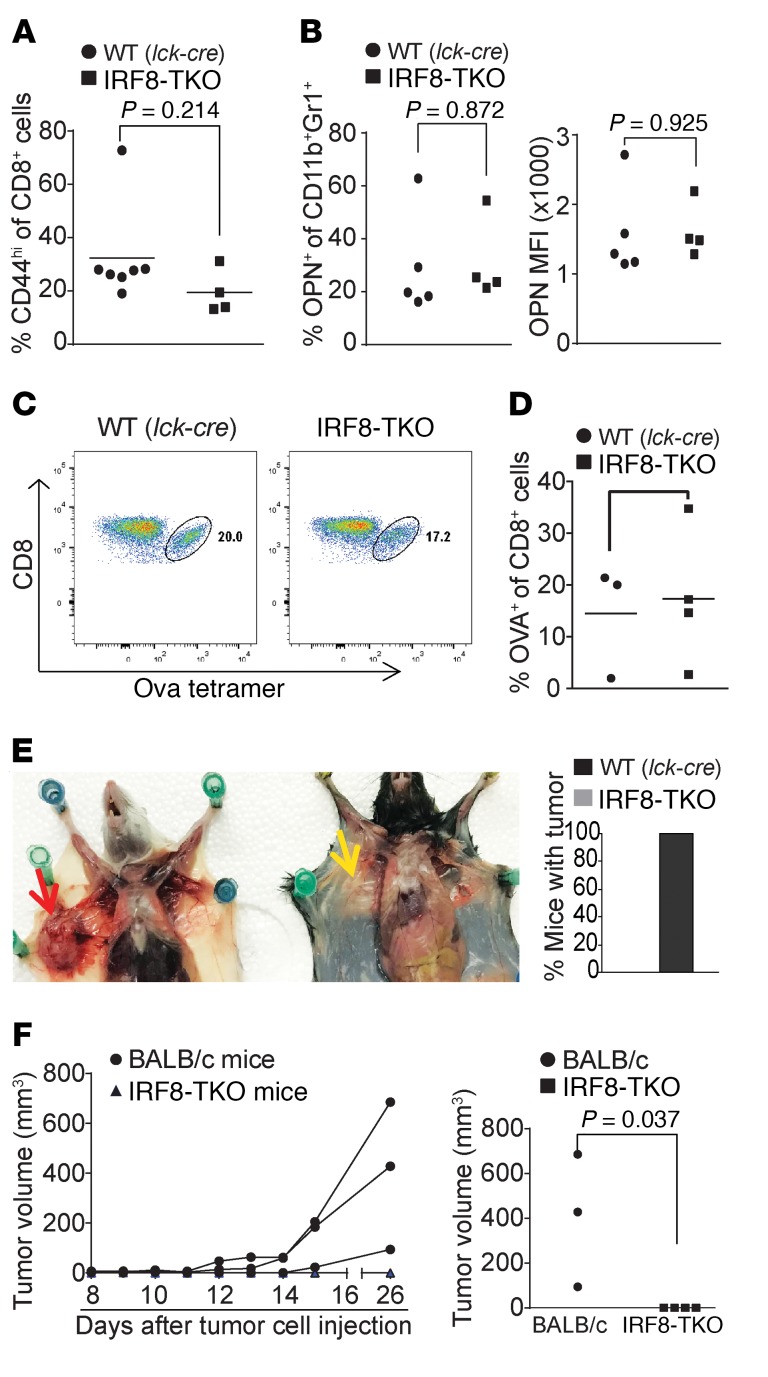
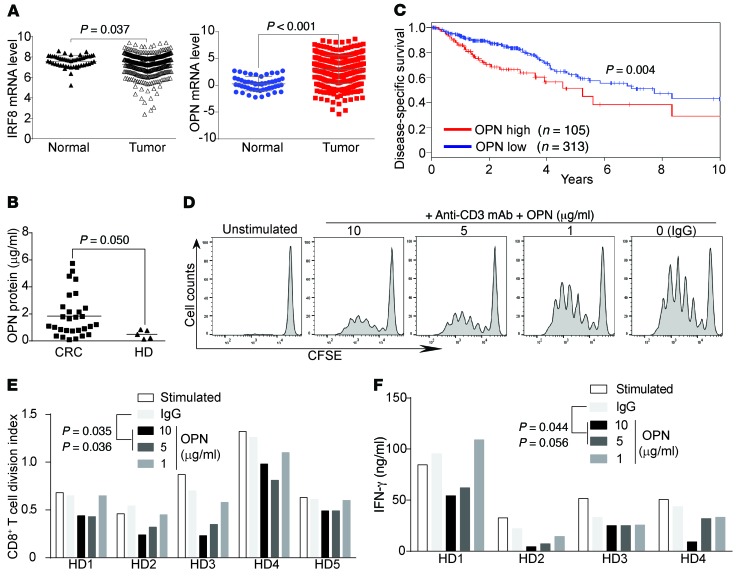
Despite breakthroughs in immune checkpoint inhibitor (ICI) immunotherapy, not all human cancers respond to ICI immunotherapy and a large fraction of patients with the responsive types of cancers do not respond to current ICI immunotherapy. This clinical conundrum suggests that additional immune checkpoints exist. We report here that interferon regulatory factor 8 (IRF8) deficiency led to impairment of cytotoxic T lymphocyte (CTL) activation and allograft tumor tolerance. However, analysis of chimera mice with competitive reconstitution of WT and IRF8-KO bone marrow cells as well as mice with IRF8 deficiency only in T cells indicated that IRF8 plays no intrinsic role in CTL activation. Instead, IRF8 functioned as a repressor of osteopontin (OPN), the physiological ligand for CD44 on T cells, in CD11b+Ly6CloLy6G+ myeloid cells and OPN acted as a potent T cell suppressor. IRF8 bound to the Spp1 promoter to repress OPN expression in colon epithelial cells, and colon carcinoma exhibited decreased IRF8 and increased OPN expression. The elevated expression of OPN in human colon carcinoma was correlated with decreased patient survival. Our data indicate that myeloid and tumor cell-expressed OPN acts as an immune checkpoint to suppress T cell activation and confer host tumor immune tolerance.
Keywords: Cancer immunotherapy; Immunology; Immunotherapy; T cells.
Conflict of interest statement
Figures


Comment in
-
Osteopontin controls immunosuppression in the tumor microenvironment.J Clin Invest. 2018 Dec 3;128(12):5209-5212. doi: 10.1172/JCI124918. Epub 2018 Nov 5. J Clin Invest. 2018. PMID: 30395537 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
