

PD-1 immunobiology in systemic lupus erythematosus
- PMID: 30396745
- PMCID: PMC7449827
- DOI: 10.1016/j.jaut.2018.10.025
PD-1 immunobiology in systemic lupus erythematosus
Abstract
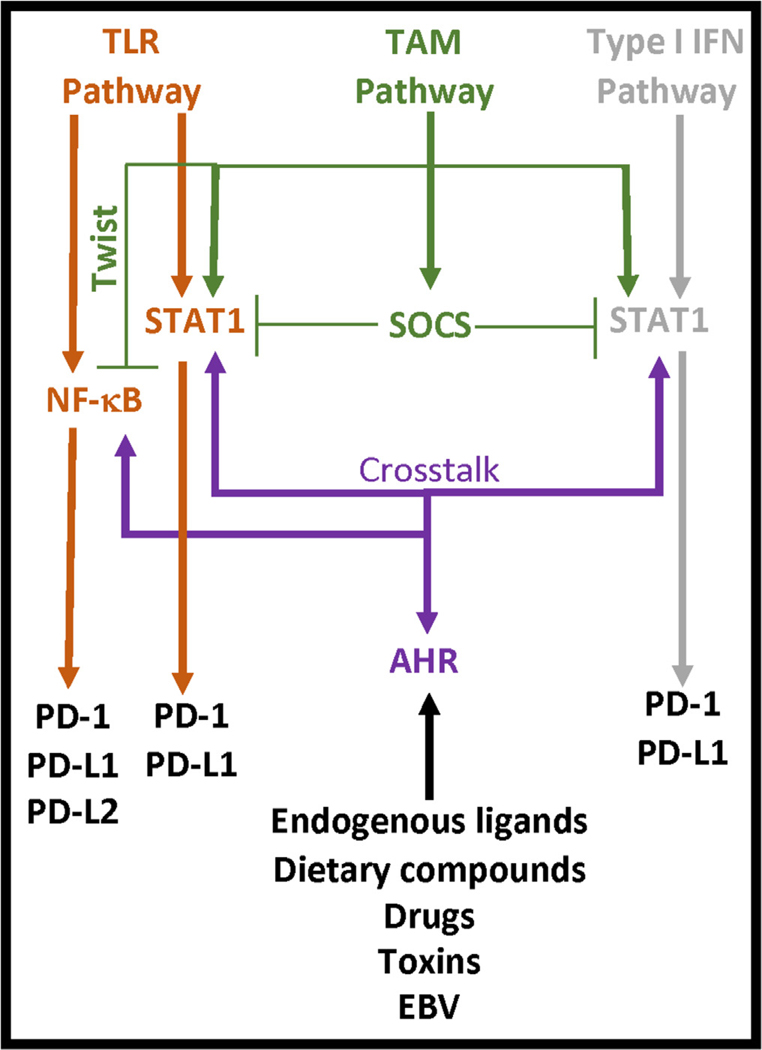
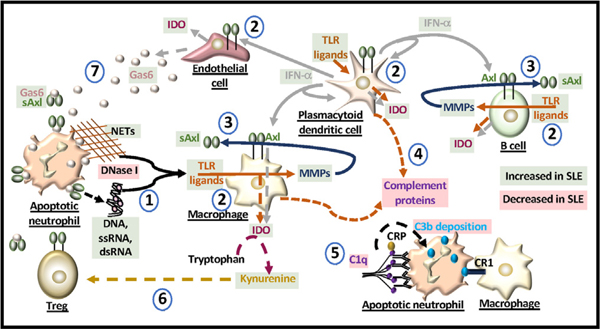
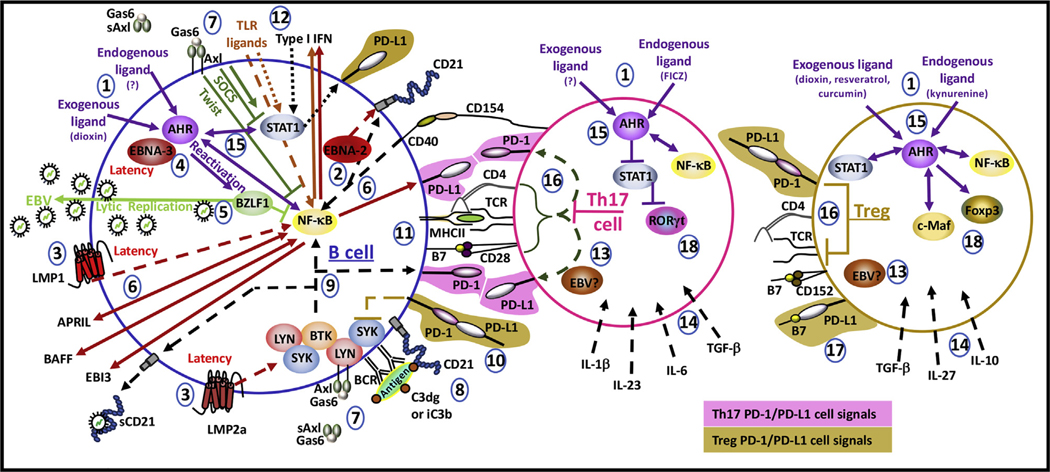
Programmed death (PD)-1 receptors and their ligands have been identified in the pathogenesis and development of systemic lupus erythematosus (SLE). Two key pathways, toll-like receptor and type I interferon, are significant to SLE pathogenesis and modulate the expression of PD-1 and the ligands (PD-L1, PD-L2) through activation of NF-κB and/or STAT1. These cell signals are regulated by tyrosine kinase (Tyro, Axl, Mer) receptors (TAMs) that are aberrantly activated in SLE. STAT1 and NF-κB also exhibit crosstalk with the aryl hydrocarbon receptor (AHR). Ligands to AHR are identified in SLE etiology and pathogenesis. These ligands also regulate the activity of the Epstein-Barr virus (EBV), which is an identified factor in SLE and PD-1 immunobiology. AHR is important in the maintenance of immune tolerance and the development of distinct immune subsets, highlighting a potential role of AHR in PD-1 immunobiology. Understanding the functions of AHR ligands as well as AHR crosstalk with STAT1, NF-κB, and EBV may provide insight into disease development, the PD-1 axis and immunotherapies that target PD-1 and its ligand, PD-L1.
Keywords: Aryl hydrocarbon receptor; Epstein-Barr virus; PD-1; Systemic lupus erythematosus.
Copyright © 2018. Published by Elsevier Ltd.
Figures
References
-
- Lee YH, Woo JH, Choi SJ, Ji JD, Song GG, Association of programmed cell death 1 polymorphisms and systemic lupus erythematosus: a meta-analysis, Lupus 18 (2009) 9–15. - PubMed
-
- Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. , Immune-related adverse events with immune checkpoint blockade: a comprehensive review, Eur. J. Cancer 54 (2016) 139–148. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
