

Early Events in the Endoplasmic Reticulum Unfolded Protein Response
- PMID: 30396883
- PMCID: PMC6442202
- DOI: 10.1101/cshperspect.a033894
Early Events in the Endoplasmic Reticulum Unfolded Protein Response
Erratum in
-
Erratum: Early Events in the Endoplasmic Reticulum Unfolded Protein Response.Cold Spring Harb Perspect Biol. 2018 Dec 3;10(12):a037309. doi: 10.1101/cshperspect.a037309. Cold Spring Harb Perspect Biol. 2018. PMID: 30510064 Free PMC article. No abstract available.
Abstract
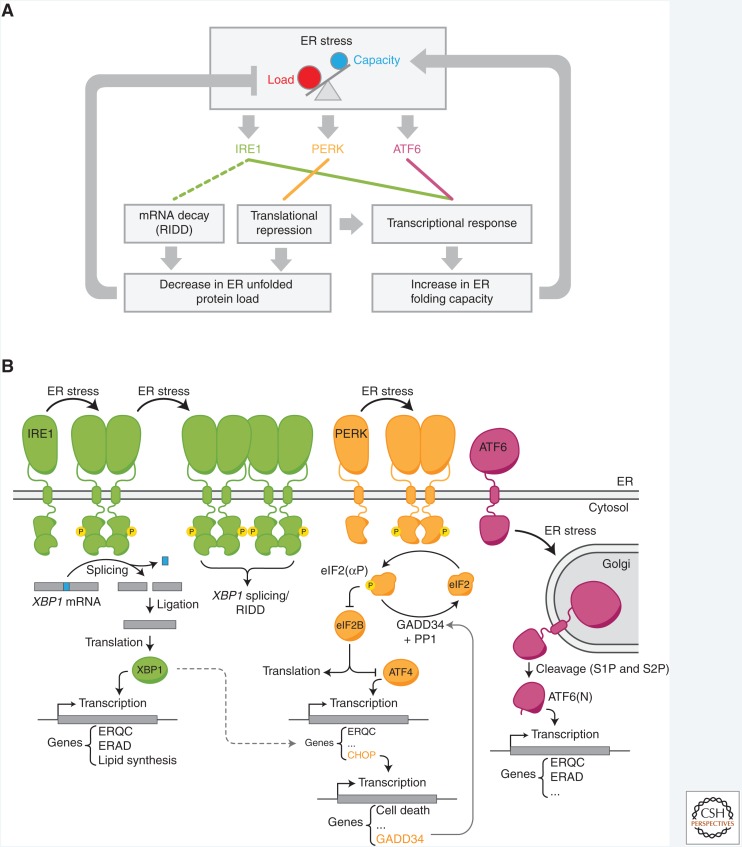
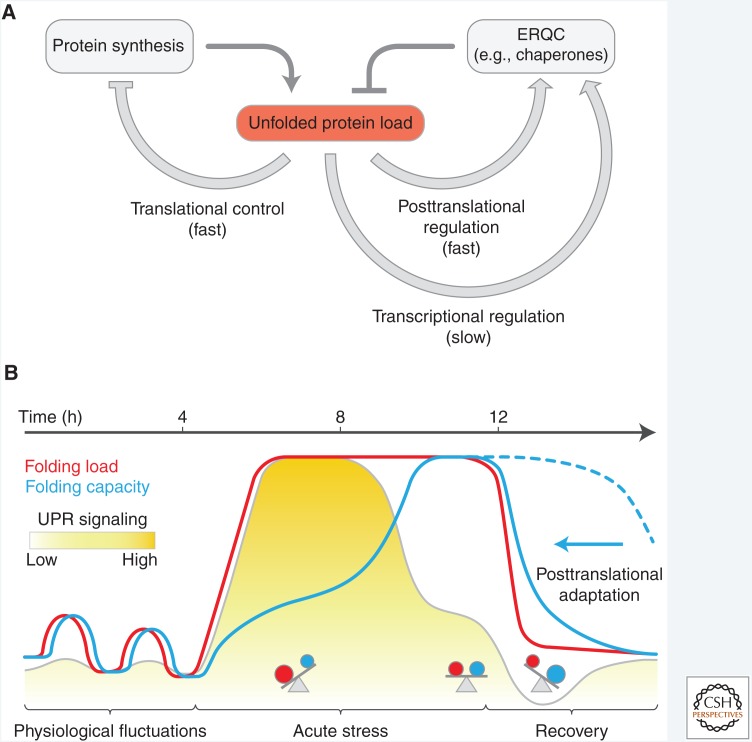
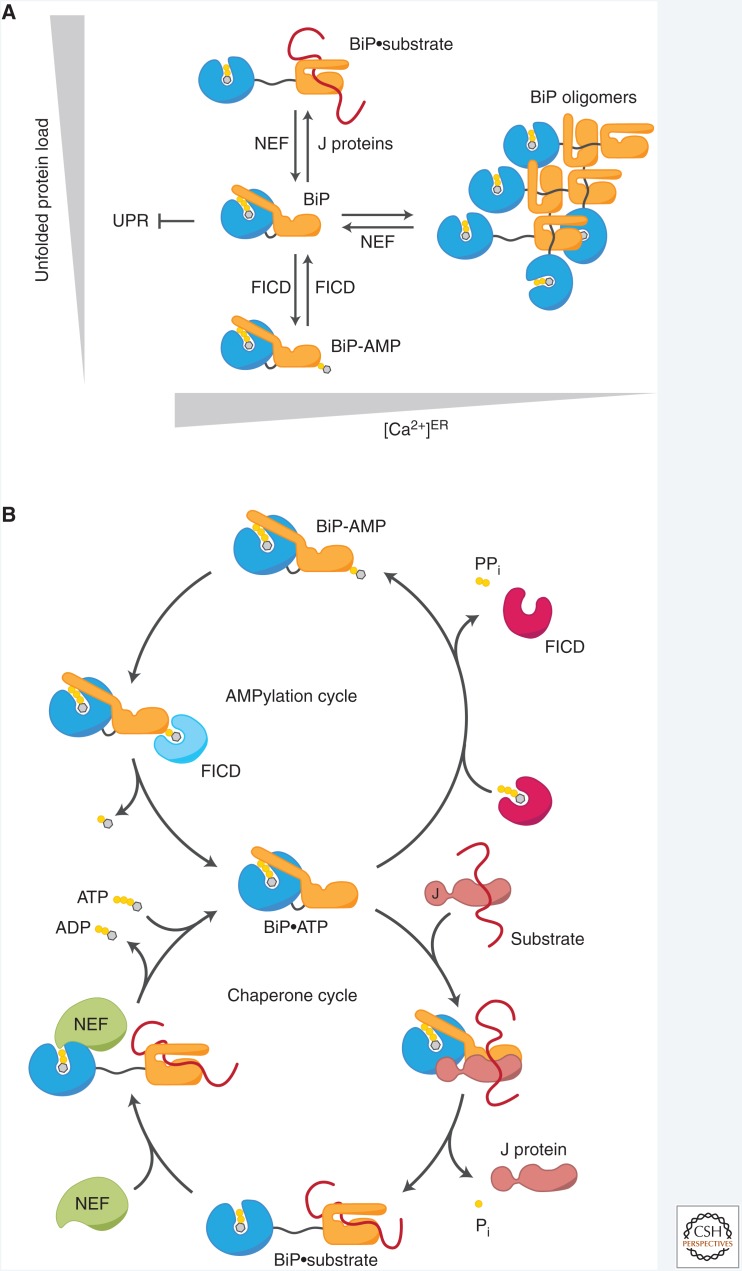
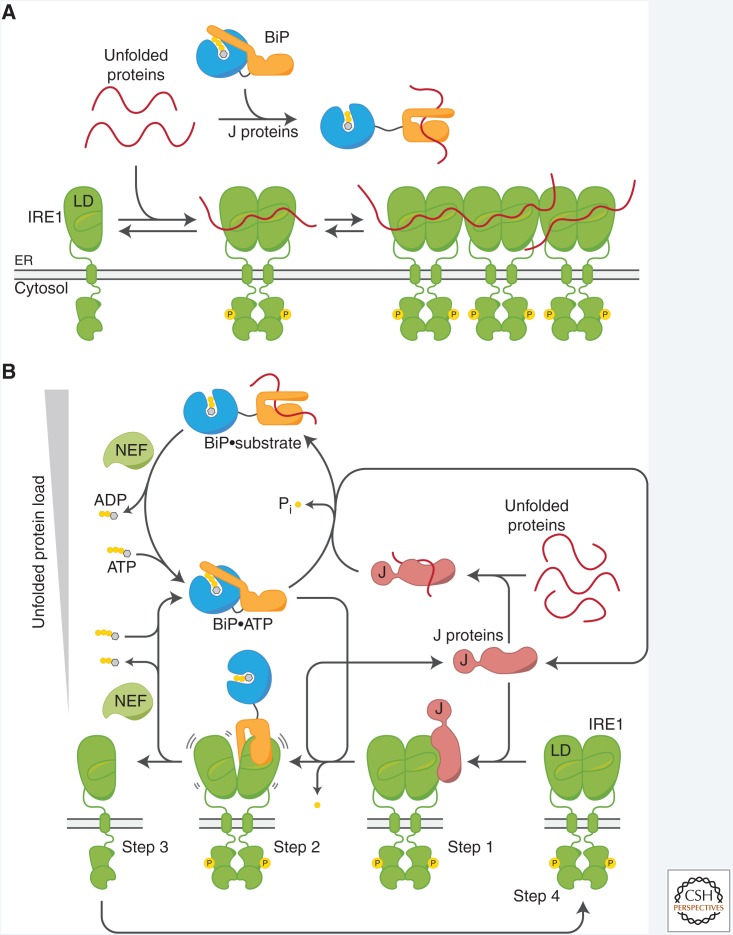
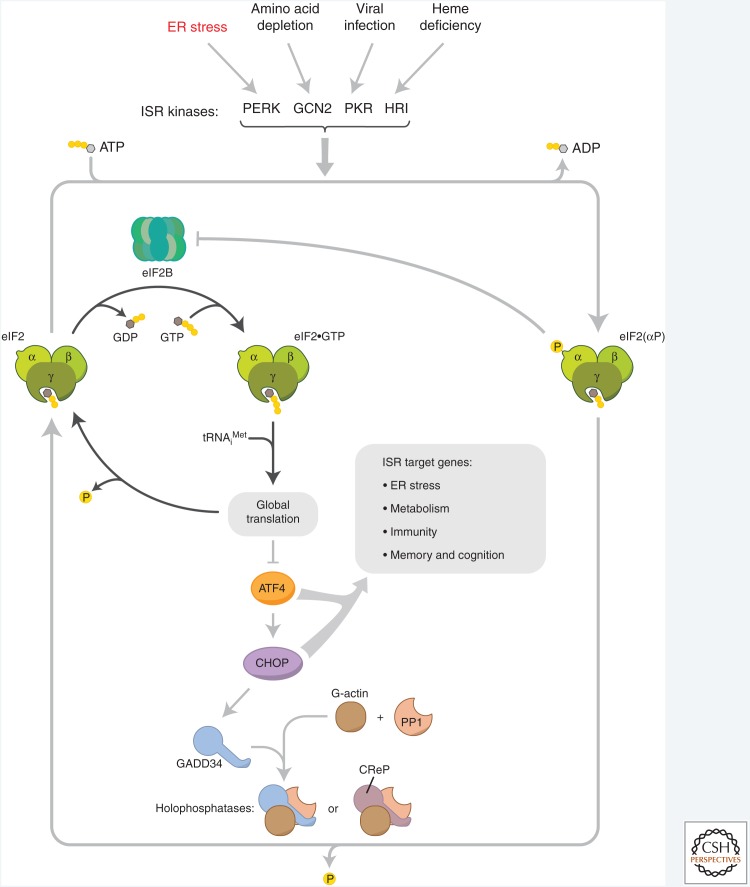
The physiological consequences of the unfolded protein response (UPR) are mediated by changes in gene expression. Underlying them are rapid processes involving preexisting components. We review recent insights gained into the regulation of the endoplasmic reticulum (ER) Hsp70 chaperone BiP, whose incorporation into inactive oligomers and reversible AMPylation and de-AMPylation present a first line of response to fluctuating levels of unfolded proteins. BiP activity is tied to the regulation of the UPR transducers by a recently discovered cycle of ER-localized, J protein-mediated formation of a repressive IRE1-BiP complex, whose working we contrast to an alternative model for UPR regulation that relies on direct recognition of unfolded proteins. We conclude with a discussion of mechanisms that repress messenger RNA (mRNA) translation to limit the flux of newly synthesized proteins into the ER, a rapid adaptation that does not rely on new macromolecule biosynthesis.
Copyright © 2019 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Abravaya K, Myers MP, Murphy SP, Morimoto RI. 1992. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev 6: 1153–1164. - PubMed
-
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et al. 2012. Discovery of 7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55: 7193–7207. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources