

CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language
- PMID: 30397230
- PMCID: PMC6218476
- DOI: 10.1038/s41467-018-06014-6
CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language
Erratum in
-
Author Correction: CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language.Nat Commun. 2019 Feb 15;10(1):883. doi: 10.1038/s41467-019-08800-2. Nat Commun. 2019. PMID: 30770872 Free PMC article.
-
Author Correction: CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language.Nat Commun. 2019 May 2;10(1):2079. doi: 10.1038/s41467-019-10161-9. Nat Commun. 2019. PMID: 31048695 Free PMC article.
Abstract
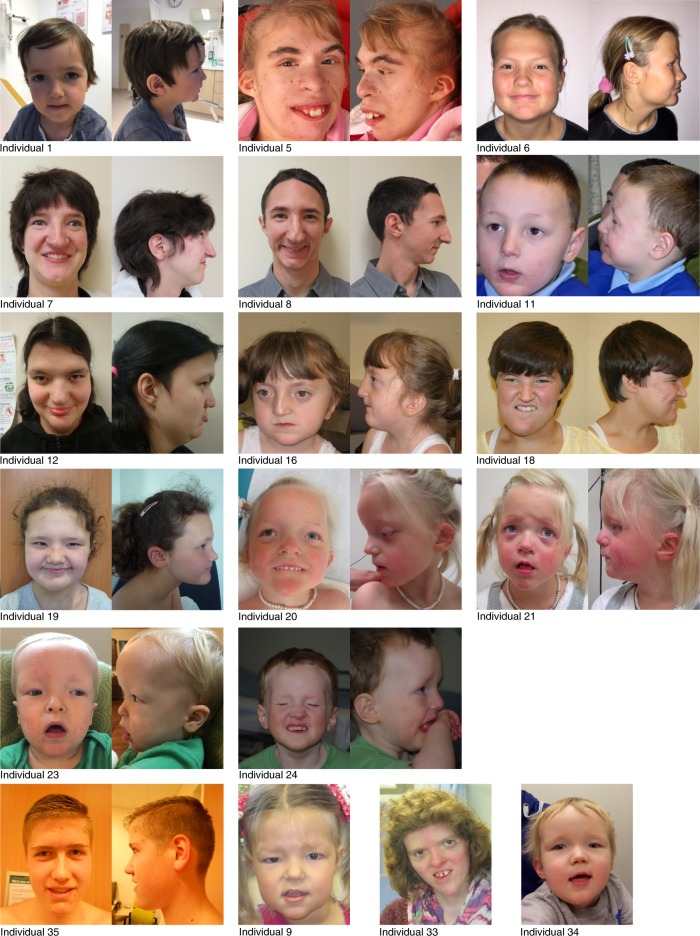
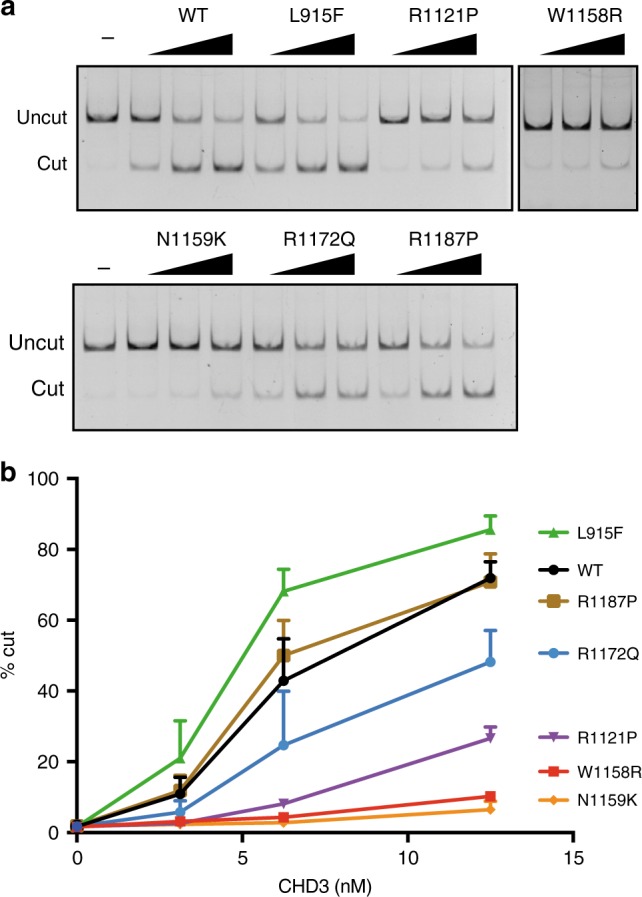
Chromatin remodeling is of crucial importance during brain development. Pathogenic alterations of several chromatin remodeling ATPases have been implicated in neurodevelopmental disorders. We describe an index case with a de novo missense mutation in CHD3, identified during whole genome sequencing of a cohort of children with rare speech disorders. To gain a comprehensive view of features associated with disruption of this gene, we use a genotype-driven approach, collecting and characterizing 35 individuals with de novo CHD3 mutations and overlapping phenotypes. Most mutations cluster within the ATPase/helicase domain of the encoded protein. Modeling their impact on the three-dimensional structure demonstrates disturbance of critical binding and interaction motifs. Experimental assays with six of the identified mutations show that a subset directly affects ATPase activity, and all but one yield alterations in chromatin remodeling. We implicate de novo CHD3 mutations in a syndrome characterized by intellectual disability, macrocephaly, and impaired speech and language.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
