

Transcriptional Landscape of PARs in Epithelial Malignancies
- PMID: 30400241
- PMCID: PMC6275037
- DOI: 10.3390/ijms19113451
Transcriptional Landscape of PARs in Epithelial Malignancies
Abstract
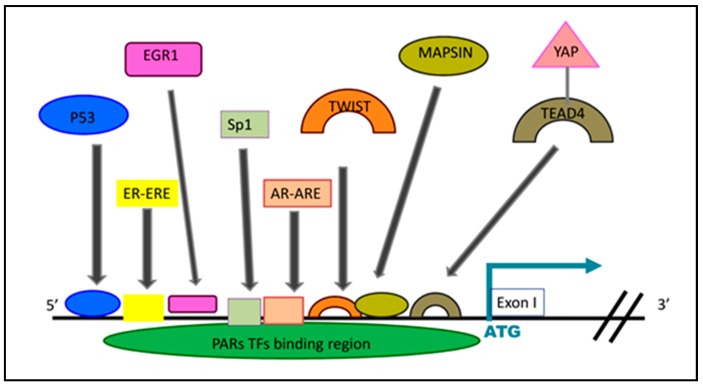
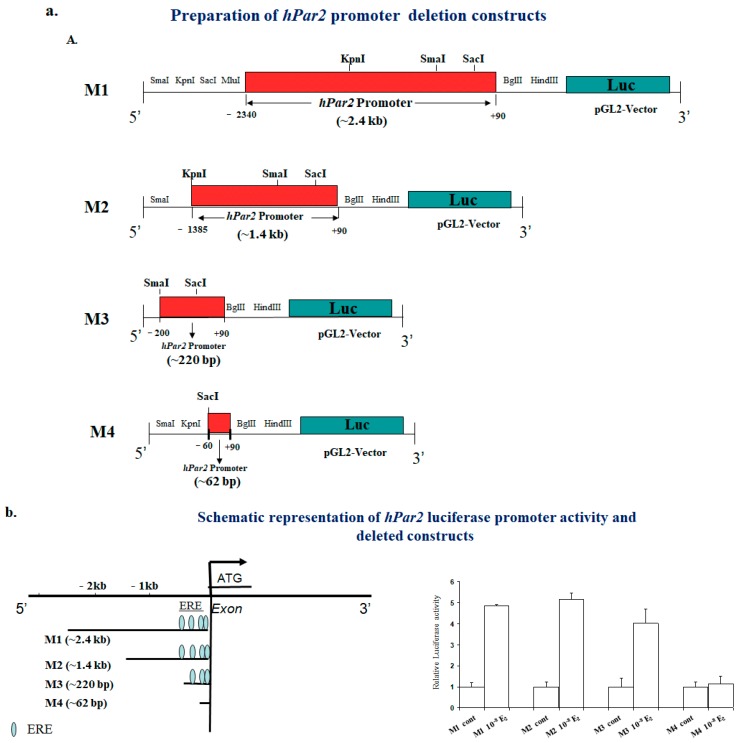
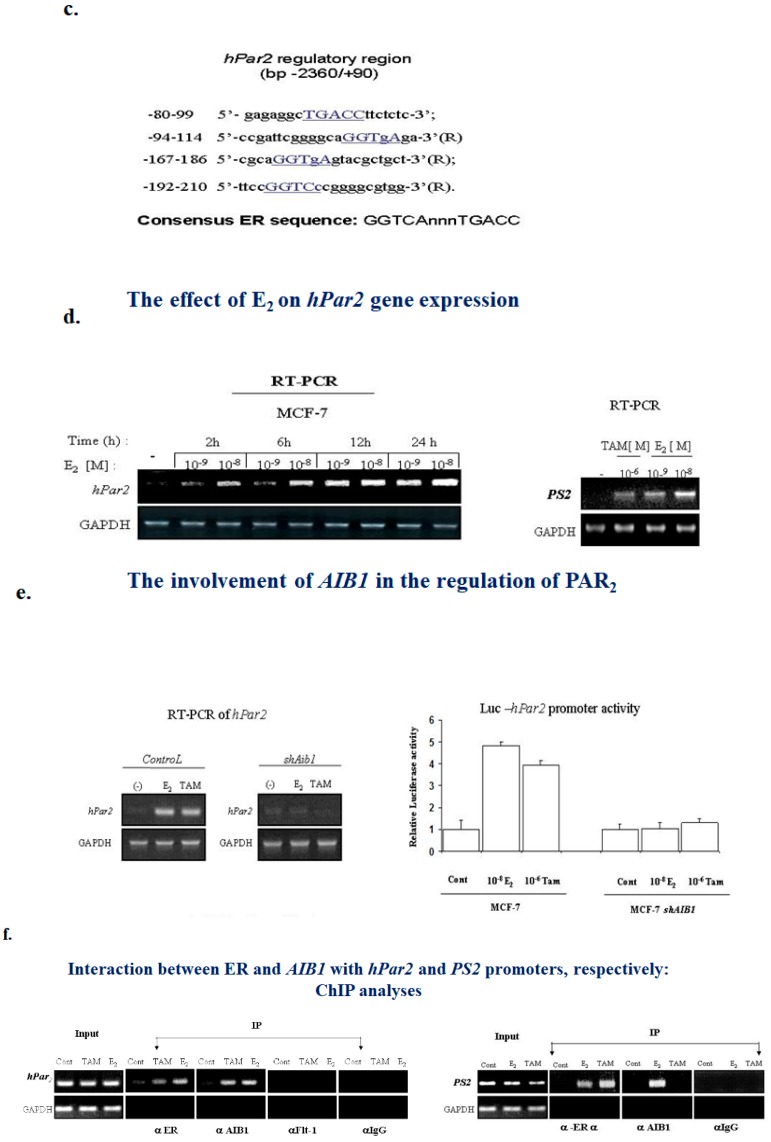
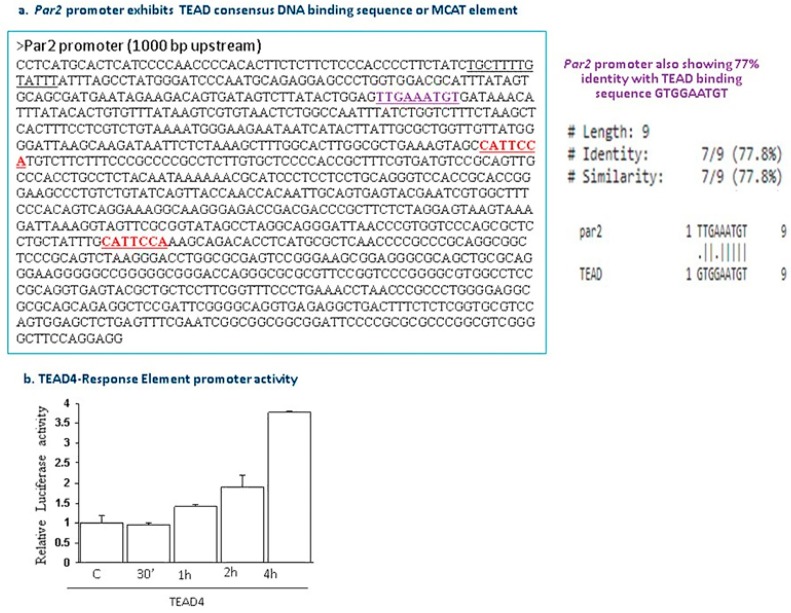
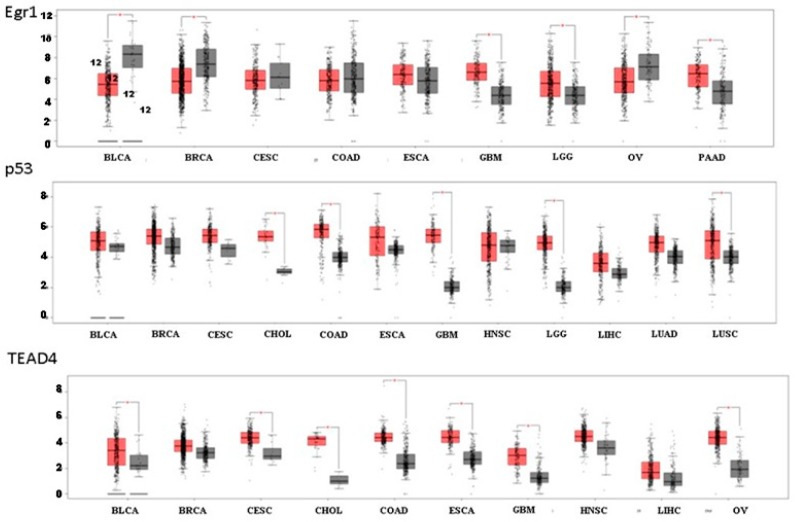
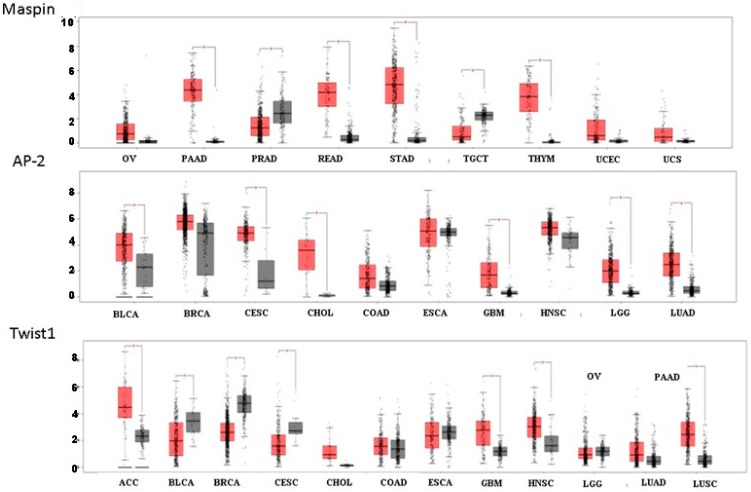
G protein-coupled receptors (GPCRs), the largest family of cell receptors, act as important regulators of diverse signaling pathways. Our understanding of the impact of GPCRs in tumors is emerging, yet there is no therapeutic platform based on GPCR driver genes. As cancer progresses, it disrupts normal epithelial organization and maintains the cells outside their normal niche. The dynamic and flexible microenvironment of a tumor contains both soluble and matrix-immobilized proteases that contribute to the process of cancer advancement. An example is the activation of cell surface protease-activated receptors (PARs). Mammalian PARs are a subgroup of GPCRs that form a family of four members, PAR1⁻4, which are uniquely activated by proteases found in the microenvironment. PAR₁ and PAR₂ play central roles in tumor biology, and PAR₃ acts as a coreceptor. The significance of PAR₄ in neoplasia is just beginning to emerge. PAR₁ has been shown to be overexpressed in malignant epithelia, in direct correlation with tumor aggressiveness, but there is no expression in normal epithelium. In this review, the involvement of key transcription factors such as Egr1, p53, Twist, AP2, and Sp1 that control PAR₁ expression levels specifically, as well as hormone transcriptional regulation by both estrogen receptors (ER) and androgen receptors (AR) are discussed. The cloning of the human protease-activated receptor 2; Par2 (hPar2) promoter region and transcriptional regulation of estrogen (E₂) via binding of the E₂⁻ER complex to estrogen response elements (ERE) are shown. In addition, evidence that TEA domain 4 (TEAD₄) motifs are present within the hPar2 promoter is presented since the YAP oncogene, which plays a central part in tumor etiology, acts via the TEAD₄ transcription factor. As of now, no information is available on regulation of the hPar3 promoter. With regard to hPar4, only data showing CpG methylation promoter regulation is available. Characterization of the PAR transcriptional landscape may identify powerful targets for cancer therapies.
Keywords: AP-2; ARE; EGR-1; ERE; PARs; Sp1; TEAD4; Twist; p53.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Combination analysis of activator protein-1 family members, Sp1 and an activator protein-2alpha-related factor binding to different regions of the urokinase receptor gene in resected colorectal cancers.Clin Cancer Res. 2005 Dec 15;11(24 Pt 1):8538-48. doi: 10.1158/1078-0432.CCR-05-0786. Clin Cancer Res. 2005. PMID: 16361535
-
GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling.Int J Mol Sci. 2018 Jun 27;19(7):1886. doi: 10.3390/ijms19071886. Int J Mol Sci. 2018. PMID: 29954076 Free PMC article. Review.
-
GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network.Int J Mol Sci. 2021 Aug 20;22(16):8985. doi: 10.3390/ijms22168985. Int J Mol Sci. 2021. PMID: 34445691 Free PMC article. Review.
-
Protease-activated receptors (PARs) in cancer: Novel biased signaling and targets for therapy.Methods Cell Biol. 2016;132:341-58. doi: 10.1016/bs.mcb.2015.11.006. Epub 2015 Dec 24. Methods Cell Biol. 2016. PMID: 26928551 Review.
-
Analysis of specific transcriptional regulators as early predictors of independent prognostic relevance in resected colorectal cancer.Clin Cancer Res. 2007 Feb 15;13(4):1123-32. doi: 10.1158/1078-0432.CCR-06-1668. Clin Cancer Res. 2007. PMID: 17317820
Cited by
-
GPCRomics: An Approach to Discover GPCR Drug Targets.Trends Pharmacol Sci. 2019 Jun;40(6):378-387. doi: 10.1016/j.tips.2019.04.001. Epub 2019 May 8. Trends Pharmacol Sci. 2019. PMID: 31078319 Free PMC article. Review.
-
GPCR-Hippo Signaling in Cancer.Cells. 2019 May 8;8(5):426. doi: 10.3390/cells8050426. Cells. 2019. PMID: 31072060 Free PMC article. Review.
-
Tamoxifen induces hypercoagulation and alterations in ERα and ERβ dependent on breast cancer sub-phenotype ex vivo.Sci Rep. 2020 Nov 6;10(1):19256. doi: 10.1038/s41598-020-75779-y. Sci Rep. 2020. PMID: 33159119 Free PMC article.
-
Antiplatelet Therapy Combined with Anastrozole Induces Features of Partial EMT in Breast Cancer Cells and Fails to Mitigate Breast-Cancer Induced Hypercoagulation.Int J Mol Sci. 2021 Apr 16;22(8):4153. doi: 10.3390/ijms22084153. Int J Mol Sci. 2021. PMID: 33923802 Free PMC article.
-
Friend or foe, the role of EGR-1 in cancer.Med Oncol. 2019 Nov 20;37(1):7. doi: 10.1007/s12032-019-1333-6. Med Oncol. 2019. PMID: 31748910 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
