

Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection
- PMID: 30400818
- PMCID: PMC6219177
- DOI: 10.1186/s13059-018-1539-5
Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection
Abstract
Background: The phenotypes of cancer cells are driven in part by somatic structural variants. Structural variants can initiate tumors, enhance their aggressiveness, and provide unique therapeutic opportunities. Whole-genome sequencing of tumors can allow exhaustive identification of the specific structural variants present in an individual cancer, facilitating both clinical diagnostics and the discovery of novel mutagenic mechanisms. A plethora of somatic structural variant detection algorithms have been created to enable these discoveries; however, there are no systematic benchmarks of them. Rigorous performance evaluation of somatic structural variant detection methods has been challenged by the lack of gold standards, extensive resource requirements, and difficulties arising from the need to share personal genomic information.
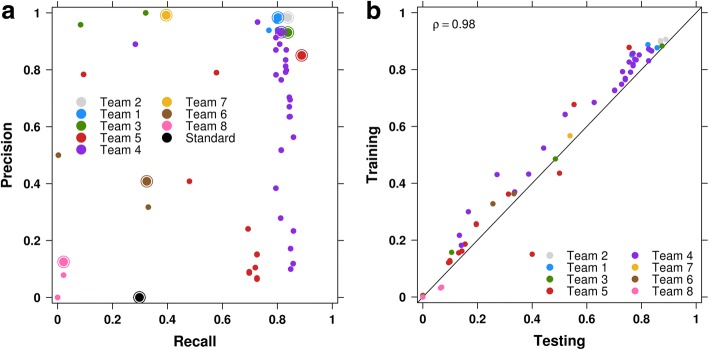
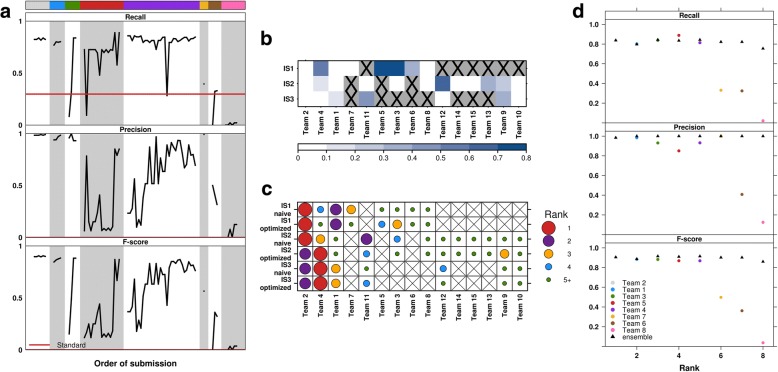
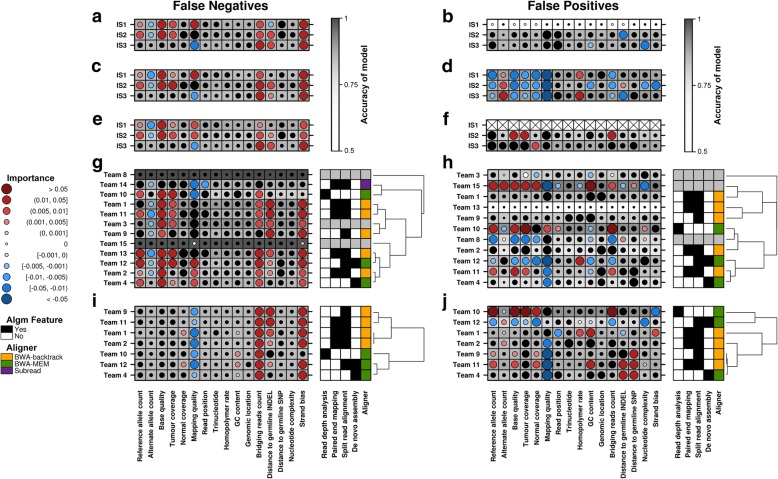
Results: To facilitate structural variant detection algorithm evaluations, we create a robust simulation framework for somatic structural variants by extending the BAMSurgeon algorithm. We then organize and enable a crowdsourced benchmarking within the ICGC-TCGA DREAM Somatic Mutation Calling Challenge (SMC-DNA). We report here the results of structural variant benchmarking on three different tumors, comprising 204 submissions from 15 teams. In addition to ranking methods, we identify characteristic error profiles of individual algorithms and general trends across them. Surprisingly, we find that ensembles of analysis pipelines do not always outperform the best individual method, indicating a need for new ways to aggregate somatic structural variant detection approaches.
Conclusions: The synthetic tumors and somatic structural variant detection leaderboards remain available as a community benchmarking resource, and BAMSurgeon is available at https://github.com/adamewing/bamsurgeon .
Keywords: Benchmarking; Cancer genomics; Crowdsourcing; Simulation; Somatic mutations; Structural variants; Whole-genome sequencing.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Huang M, Ye Y, Chen S, Chai J, Lu J, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–572. - PubMed
-
- Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–1532. doi: 10.1016/S1470-2045(14)71021-6. - DOI - PubMed
