

Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation
- PMID: 30401786
- PMCID: PMC6329291
- DOI: 10.1126/scisignal.aaq1075
Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation
Abstract
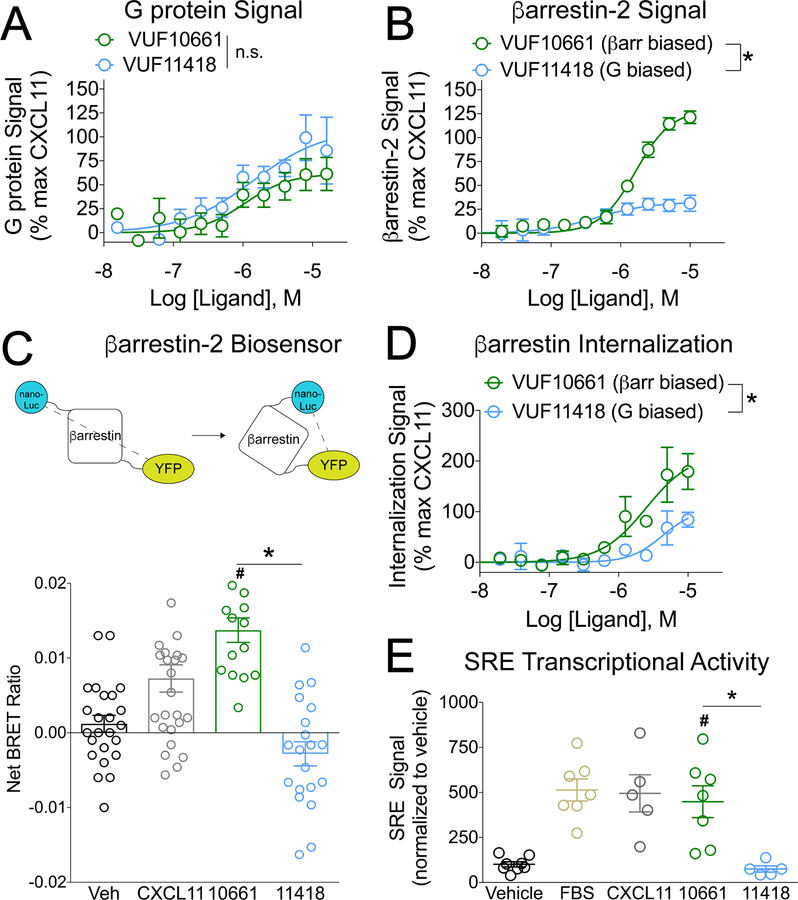
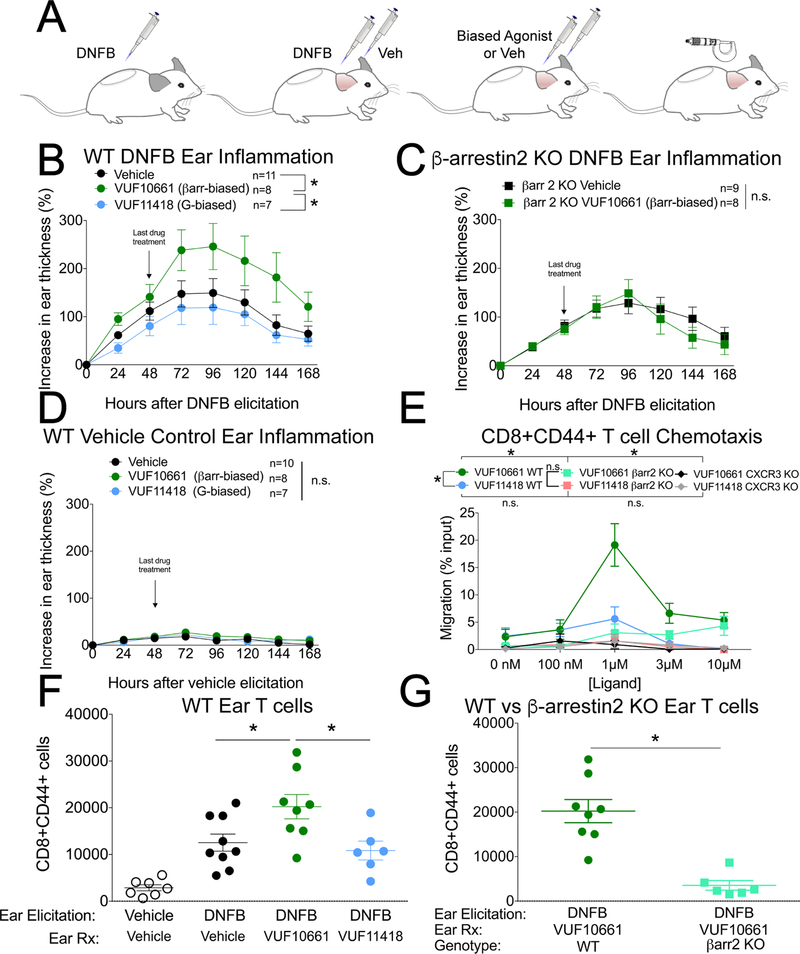
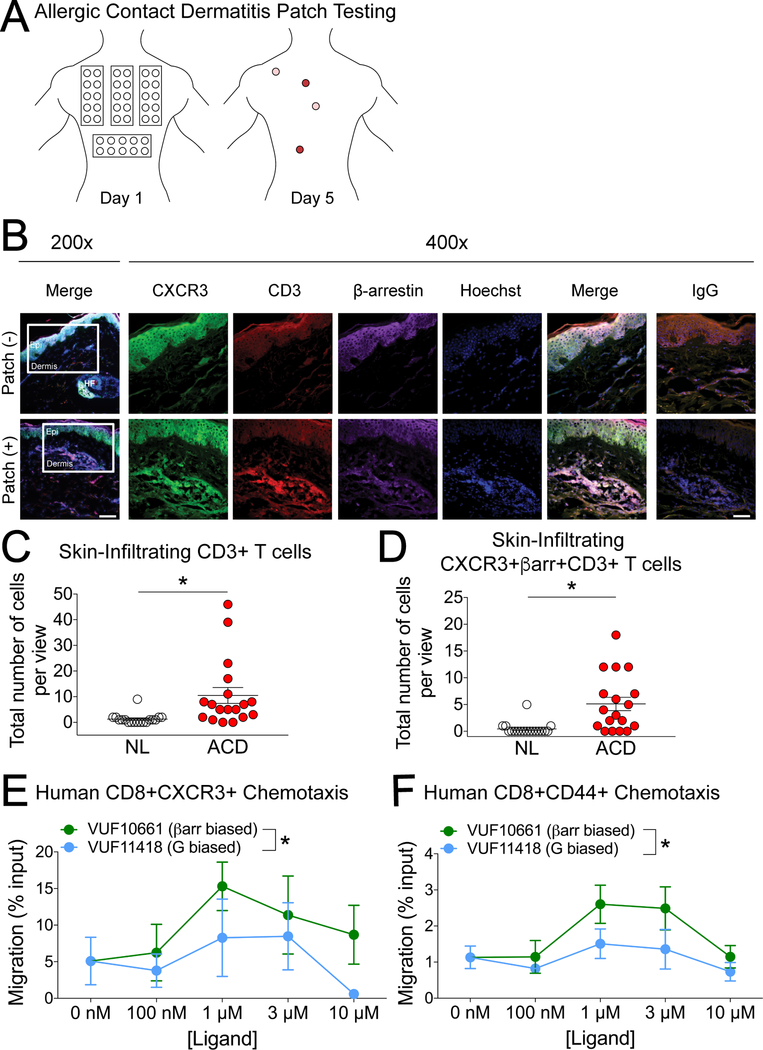
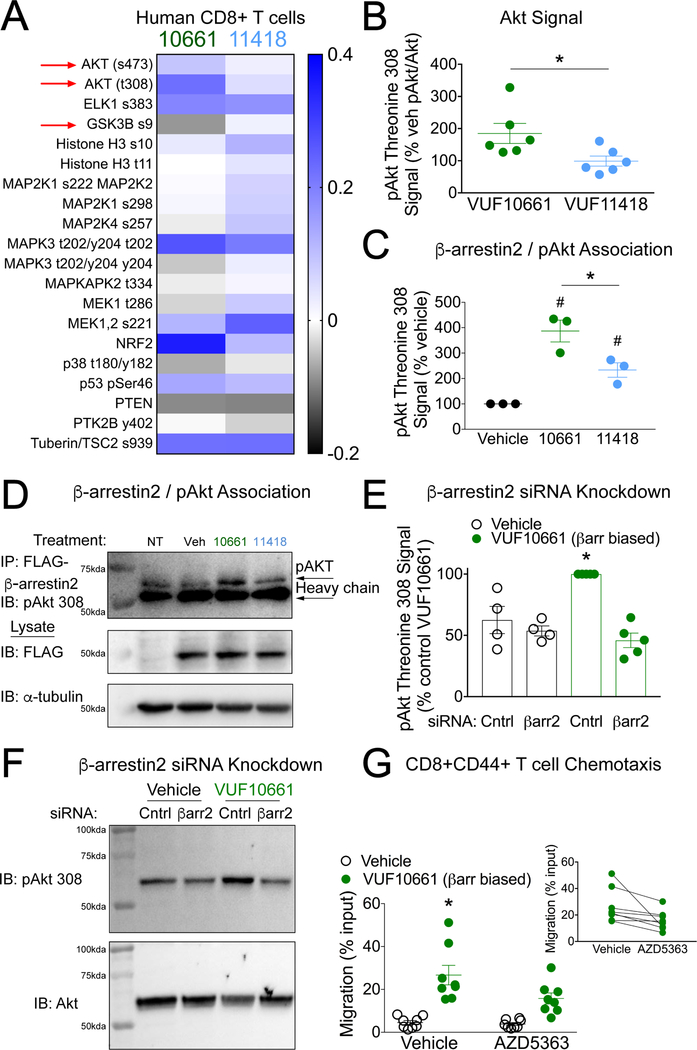
The chemokine receptor CXCR3 plays a central role in inflammation by mediating effector/memory T cell migration in various diseases; however, drugs targeting CXCR3 and other chemokine receptors are largely ineffective in treating inflammation. Chemokines, the endogenous peptide ligands of chemokine receptors, can exhibit so-called biased agonism by selectively activating either G protein- or β-arrestin-mediated signaling after receptor binding. Biased agonists might be used as more targeted therapeutics to differentially regulate physiological responses, such as immune cell migration. To test whether CXCR3-mediated physiological responses could be segregated by G protein- and β-arrestin-mediated signaling, we identified and characterized small-molecule biased agonists of the receptor. In a mouse model of T cell-mediated allergic contact hypersensitivity (CHS), topical application of a β-arrestin-biased, but not a G protein-biased, agonist potentiated inflammation. T cell recruitment was increased by the β-arrestin-biased agonist, and biopsies of patients with allergic CHS demonstrated coexpression of CXCR3 and β-arrestin in T cells. In mouse and human T cells, the β-arrestin-biased agonist was the most efficient at stimulating chemotaxis. Analysis of phosphorylated proteins in human lymphocytes showed that β-arrestin-biased signaling activated the kinase Akt, which promoted T cell migration. This study demonstrates that biased agonists of CXCR3 produce distinct physiological effects, suggesting discrete roles for different endogenous CXCR3 ligands and providing evidence that biased signaling can affect the clinical utility of drugs targeting CXCR3 and other chemokine receptors.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
-
- Schall TJ, Proudfoot AE, Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol 11, 355–363 (2011). - PubMed
-
- Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, Fujimoto K, Yamamoto K, Miyamoto T, Uike N, Tanimoto M, Tsukasaki K, Ishizawa K, Suzumiya J, Inagaki H, Tamura K, Akinaga S, Tomonaga M, Ueda R, Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol 32, 1157–1163 (2014). - PubMed
-
- Mantovani A, The chemokine system: redundancy for robust outputs. Immunol Today 20, 254–257 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
