

miR-26a Potentially Contributes to the Regulation of Fatty Acid and Sterol Metabolism In Vitro Human HepG2 Cell Model of Nonalcoholic Fatty Liver Disease
- PMID: 30402207
- PMCID: PMC6196797
- DOI: 10.1155/2018/8515343
miR-26a Potentially Contributes to the Regulation of Fatty Acid and Sterol Metabolism In Vitro Human HepG2 Cell Model of Nonalcoholic Fatty Liver Disease
Abstract
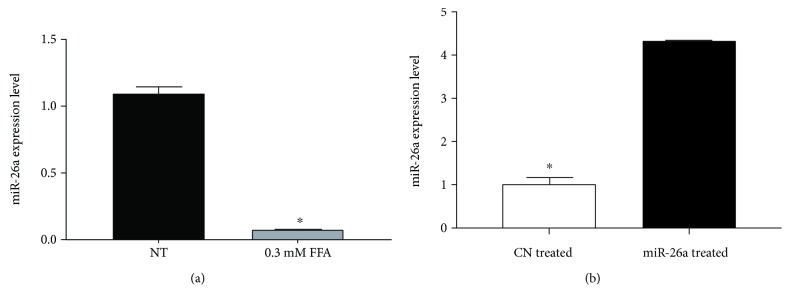
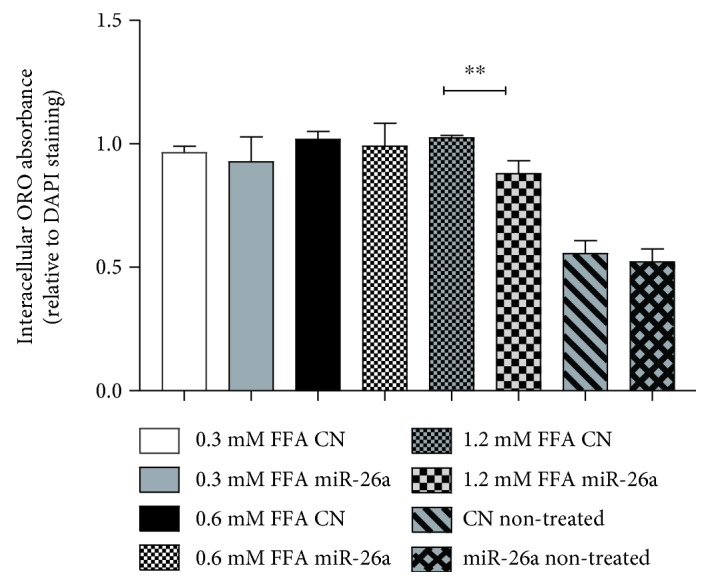
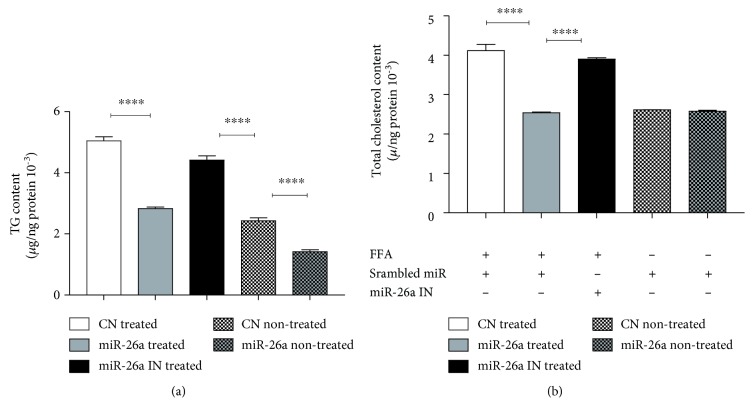
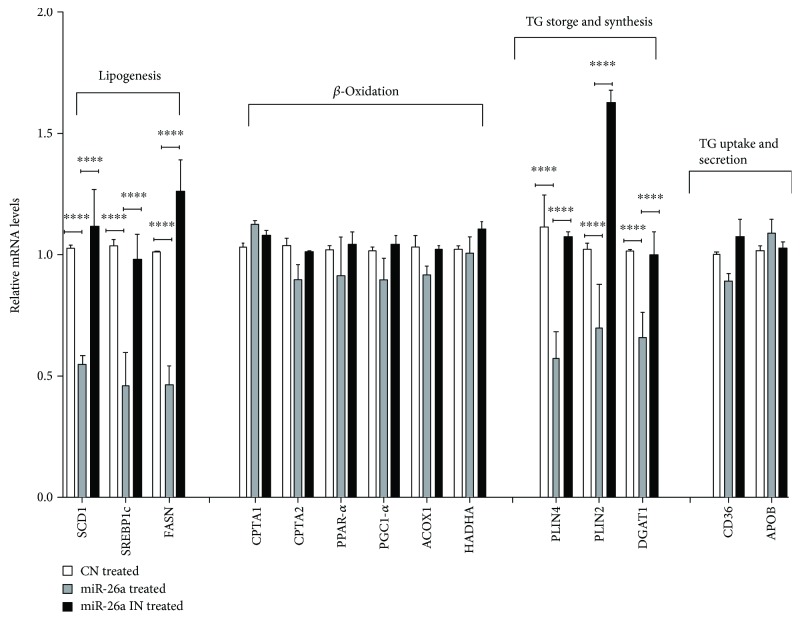
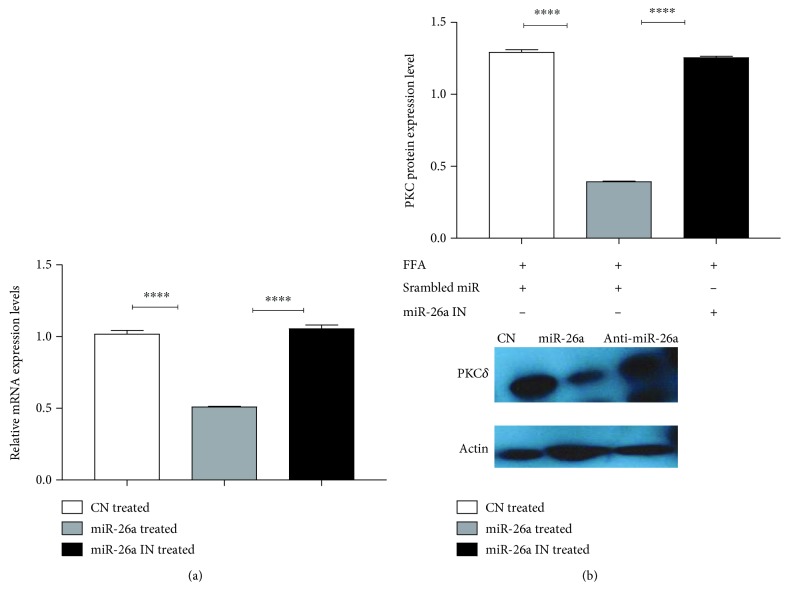
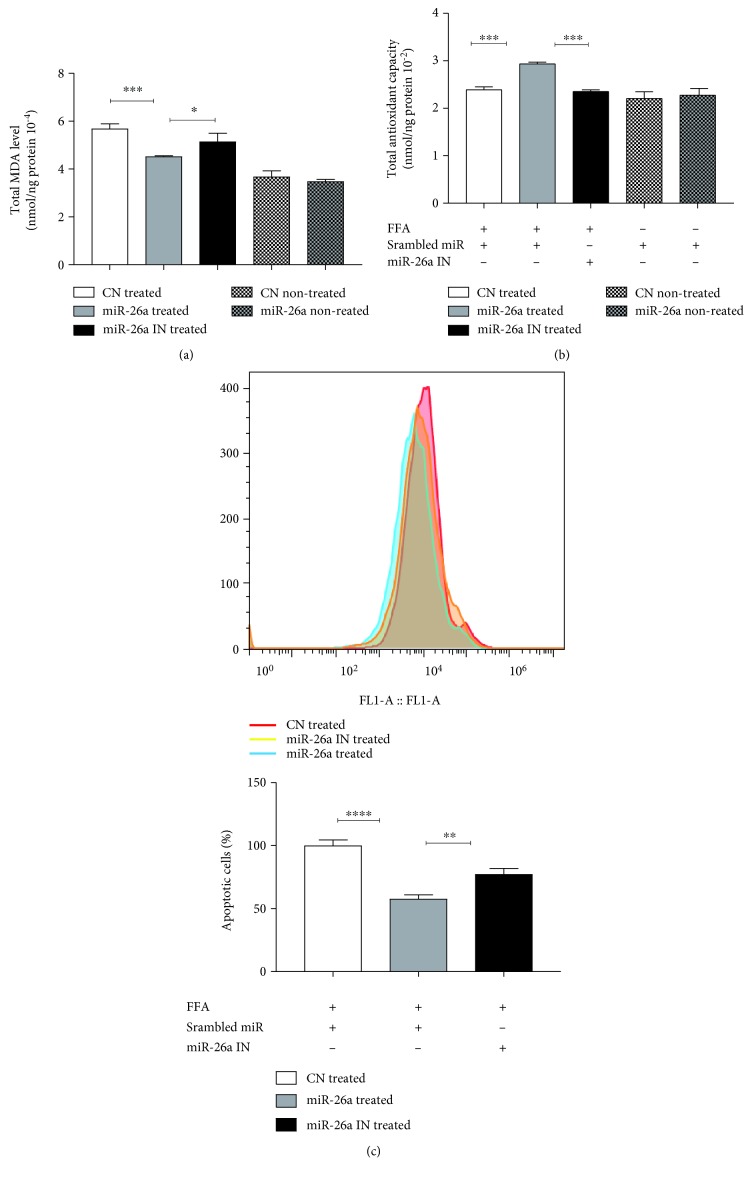
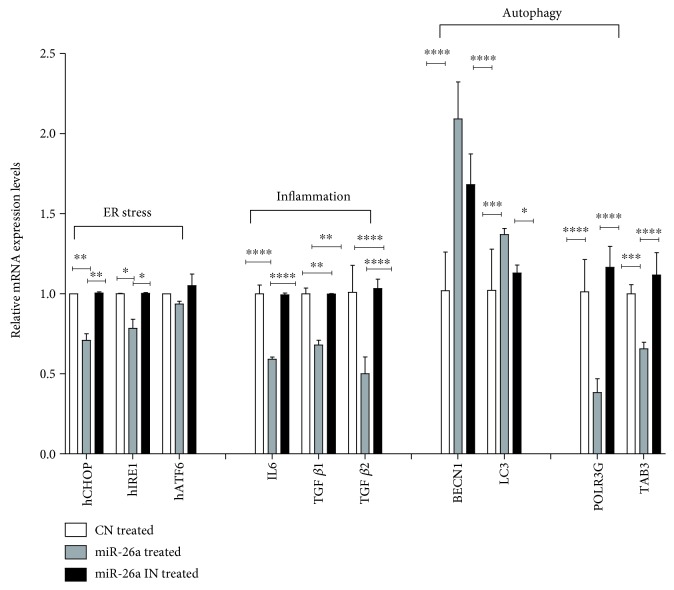
Nonalcoholic fatty liver disease (NAFLD) is a metabolic-related disorder ranging from steatosis to steatohepatitis, which may progress to cirrhosis and hepatocellular carcinoma (HCC). This study aimed at assessing the regulatory and protective role of miR-26a on lipid metabolism and progression of NAFLD in human HepG2 cells loaded with free fatty acids (FFA). Lentivirus expressing miR-26a or negative control miR was used to transduce HepG2 cells and to establish stable cell lines. Gain or loss of function using an miR-26a inhibitor was used to compare triglyceride content (TG), total cholesterol level (CL), total antioxidant capacity (TAC), malondialdehyde (MDA) and the level of apoptosis. In addition, quantitative reverse transcription polymerase chain reaction (qPCR) was used to assess the mRNA levels of lipogenesis, TG synthesis, storage genes, inflammatory and fibrogenic markers, and autophagic besides endoplasmic reticulum (ER) stress markers after gaining or losing the function of miR-26a. miR-26a levels decreased in response to FFA in human HepG2 cells. After the establishment of a stable cell line, the upregulation of miR-26a resulted in the downregulation of TG, CL, and MDA levels, through regulating mRNA levels of genes involved in lipid homeostasis, ER stress marker, inflammatory and fibrogenic markers. Nevertheless, there was a marked increment in the mRNA expression of autophagic marker genes. Moreover, miR-26a overexpression protects the cells from apoptosis, whereas inhibition of miR-26a, using an anti-miR-26a oligonucleotide, decreased the expression of miR-26a which potentially contributes to altered lipid metabolism in HepG2 cells loaded with FFA. In conclusion, these findings suggested that miR-26a has a crucial role in regulating fatty acid and cholesterol homeostasis in HepG2 cells, along with the offered protection against the progression of NAFLD in vitro. Hence, miRNAs could receive growing attention as useful noninvasive diagnostic markers to follow the progression of NAFLD and to identify novel therapeutic targets.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
