

Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma
- PMID: 30402543
- PMCID: PMC6209383
- DOI: 10.1126/sciadv.aau5935
Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma
Abstract
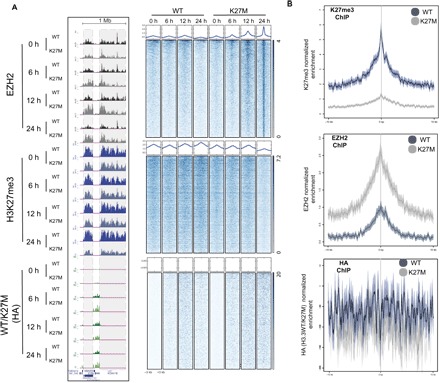
A methionine substitution at lysine-27 on histone H3 variants (H3K27M) characterizes ~80% of diffuse intrinsic pontine gliomas (DIPG) and inhibits polycomb repressive complex 2 (PRC2) in a dominant-negative fashion. Yet, the mechanisms for this inhibition and abnormal epigenomic landscape have not been resolved. Using quantitative proteomics, we discovered that robust PRC2 inhibition requires levels of H3K27M greatly exceeding those of PRC2, seen in DIPG. While PRC2 inhibition requires interaction with H3K27M, we found that this interaction on chromatin is transient, with PRC2 largely being released from H3K27M. Unexpectedly, inhibition persisted even after PRC2 dissociated from H3K27M-containing chromatin, suggesting a lasting impact on PRC2. Furthermore, allosterically activated PRC2 is particularly sensitive to H3K27M, leading to the failure to spread H3K27me from PRC2 recruitment sites and consequently abrogating PRC2's ability to establish H3K27me2-3 repressive chromatin domains. In turn, levels of polycomb antagonists such as H3K36me2 are elevated, suggesting a more global, downstream effect on the epigenome. Together, these findings reveal the conditions required for H3K27M-mediated PRC2 inhibition and reconcile seemingly paradoxical effects of H3K27M on PRC2 recruitment and activity.
Figures





References
-
- Talbert P. B., Henikoff S., Histone variants on the move: Substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol. 18, 115–126 (2017). - PubMed
-
- Jones C., Karajannis M. A., Jones D. T. W., Kieran M. W., Monje M., Baker S. J., Becher O. J., Cho Y.-J., Gupta N., Hawkins C., Hargrave D., Haas-Kogan D. A., Jabado N., Li X.-N., Mueller S., Nicolaides T., Packer R. J., Persson A. I., Phillips J. J., Simonds E. F., Stafford J. M., Tang Y., Pfister S. M., Weiss W. A., Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 19, 153–161 (2016). - PMC - PubMed
-
- Bender S., Tang Y., Lindroth A. M., Hovestadt V., Jones D. T., Kool M., Zapatka M., Northcott P. A., Sturm D., Wang W., Radlwimmer B., Højfeldt J. W., Truffaux N., Castel D., Schubert S., Ryzhova M., Seker-Cin H., Gronych J., Johann P. D., Stark S., Meyer J., Milde T., Schuhmann M., Ebinger M., Monoranu C. M., Ponnuswami A., Chen S., Jones C., Witt O., Collins V. P., von Deimling A., Jabado N., Puget S., Grill J., Helin K., Korshunov A., Lichter P., Monje M., Plass C., Cho Y. J., Pfister S. M., Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013). - PubMed
-
- Mohammad F., Weissmann S., Leblanc B., Pandey D. P., Højfeldt J. W., Comet I., Zheng C., Johansen J. V., Rapin N., Porse B. T., Tvardovskiy A., Jensen O. N., Olaciregui N. G., Lavarino C., Suñol M., de Torres C., Mora J., Carcaboso A. M., Helin K., EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
