

Chemo-Selection Strategy for Limited Proteolysis Experiments on the Proteomic Scale
- PMID: 30403842
- PMCID: PMC6524534
- DOI: 10.1021/acs.analchem.8b04122
Chemo-Selection Strategy for Limited Proteolysis Experiments on the Proteomic Scale
Abstract
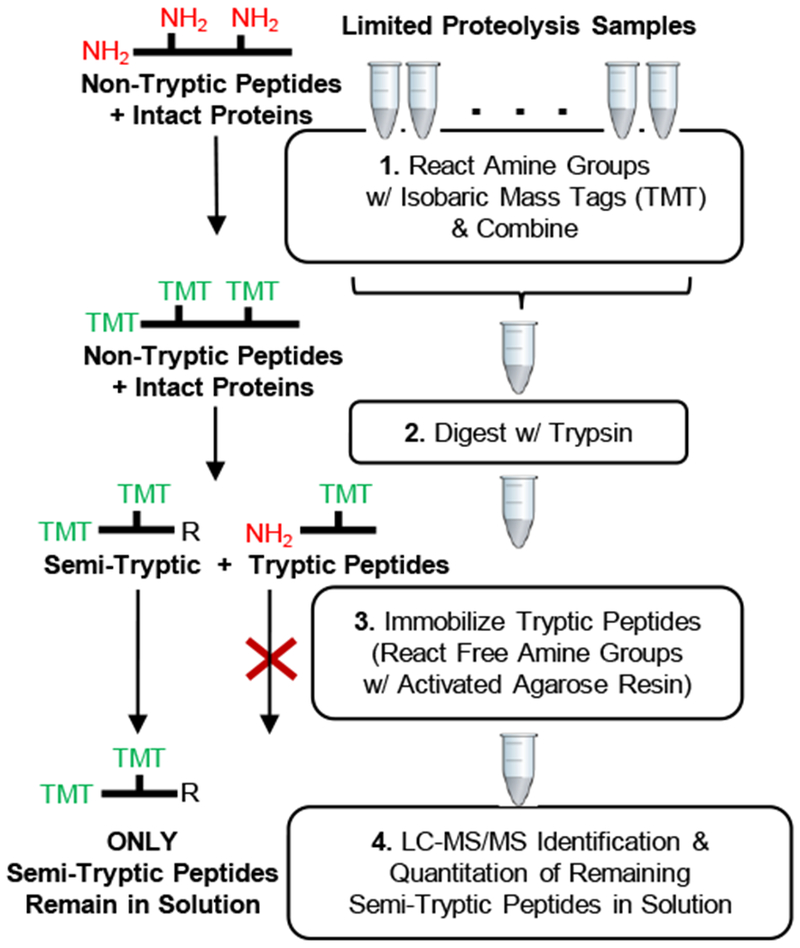
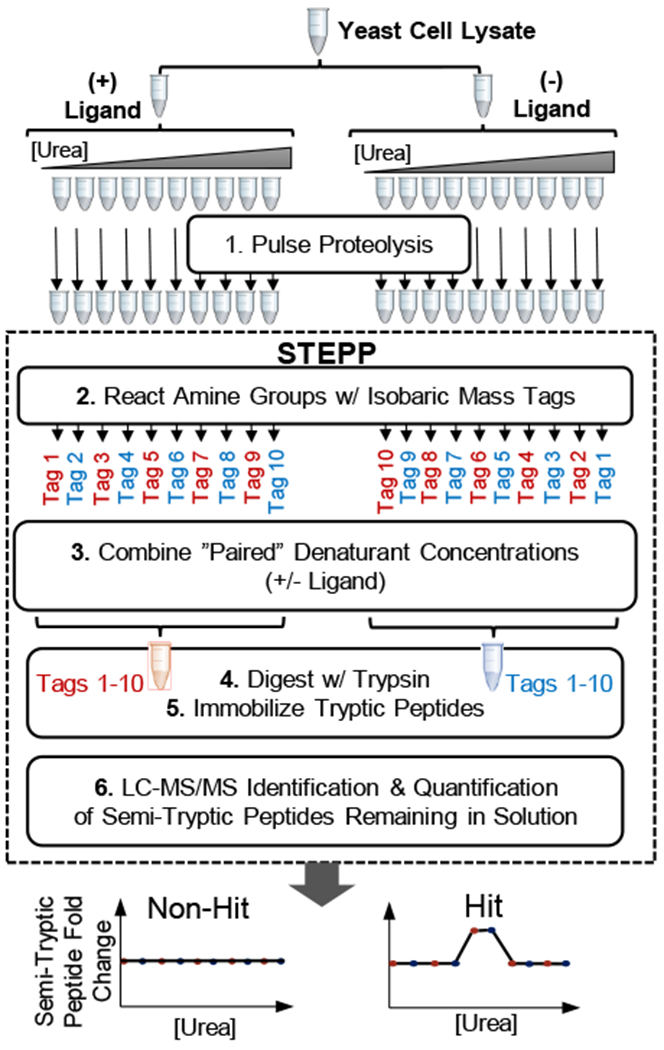
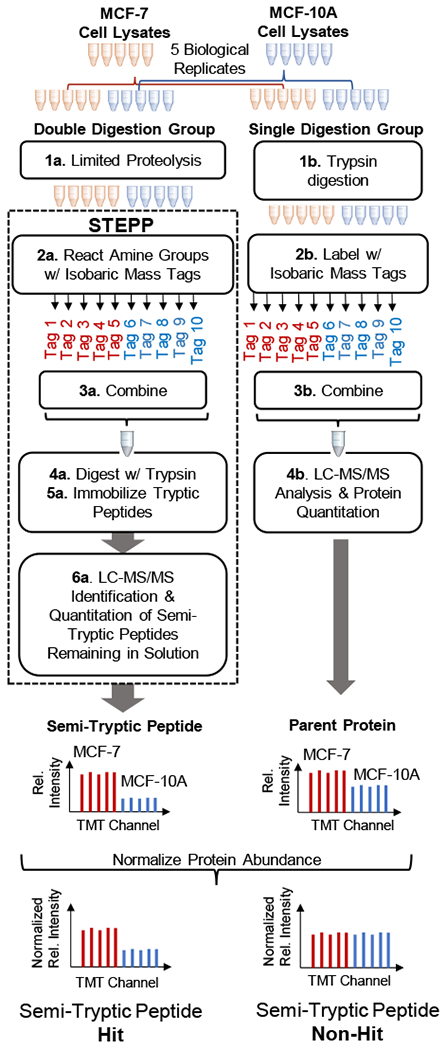
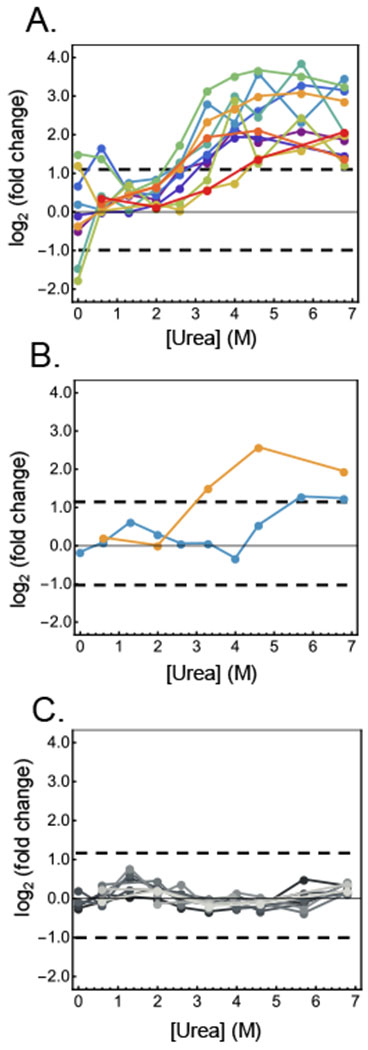
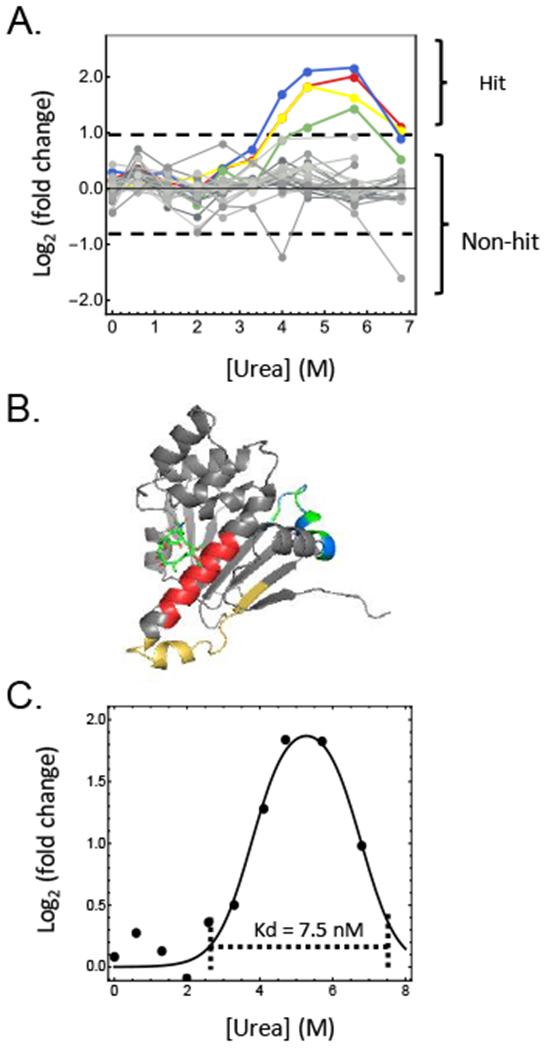
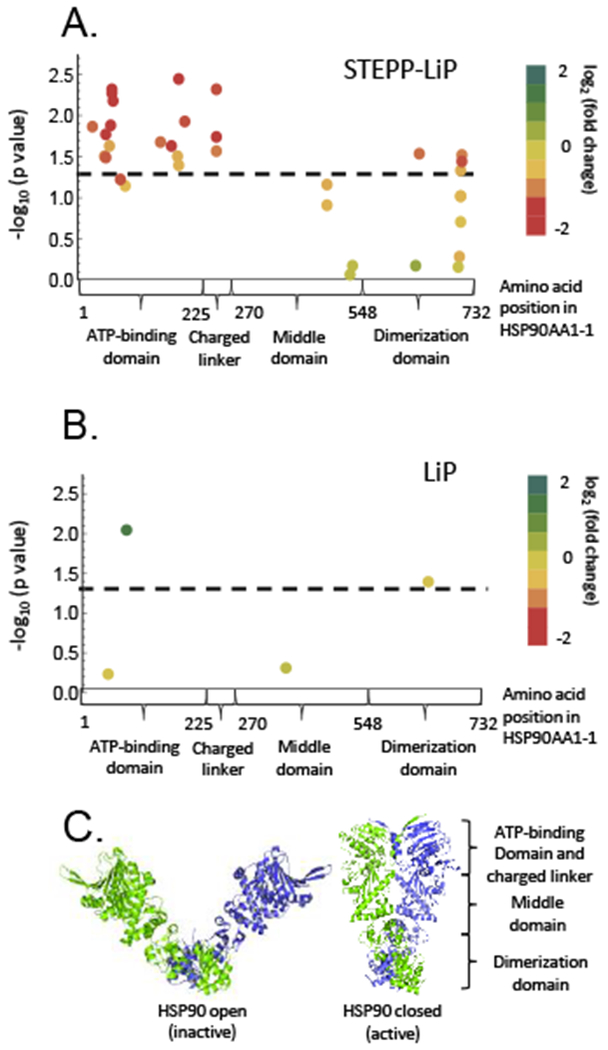
Described here is a chemo-selective enrichment strategy, termed the semitryptic peptide enrichment strategy for proteolysis procedures (STEPP), to isolate the semitryptic peptides generated in mass spectrometry-based proteome-wide applications of limited proteolysis methods. The strategy involves reacting the ε-amino groups of lysine side chains and any N-termini created in the limited proteolysis reaction with isobaric mass tags. A subsequent digestion of the sample with trypsin and the chemo-selective reaction of the newly exposed N-termini of the tryptic peptides with N-hydroxysuccinimide (NHS)-activated agarose resin removes the tryptic peptides from solution, leaving only the semitryptic peptides with one nontryptic cleavage site generated in the limited proteolysis reaction for subsequent LC-MS/MS analysis. As part of this work, the STEPP technique is interfaced with two different proteolysis methods, including the pulse proteolysis (PP) and limited proteolysis (LiP) methods. The STEPP-PP workflow is evaluated in two proof-of-principle experiments involving the proteins in a yeast cell lysate and two well-studied drugs, cyclosporin A and geldanamycin. The STEPP-LiP workflow is evaluated in a proof-of-principle experiment involving the proteins in two cell culture models of human breast cancer, MCF-7 and MCF-10A cell lines. The STEPP protocol increased the number of semitryptic peptides detected in the LiP and PP experiments by 5- to 10-fold. The STEPP protocol not only increases the proteomic coverage, but also increases the amount of structural information that can be gleaned from limited proteolysis experiments. Moreover, the protocol also enables the quantitative determination of ligand binding affinities.
Figures
Similar articles
-
Multiplexed Protease Specificity Profiling Using Isobaric Labeling.Methods Mol Biol. 2017;1574:171-182. doi: 10.1007/978-1-4939-6850-3_12. Methods Mol Biol. 2017. PMID: 28315250
-
Mimicking LysC Proteolysis by 'Arginine Modification-cum-Trypsin Digestion': Comparison of Bottom-up & Middle-down Proteomic Approaches by ESI Q-TOF MS.Protein Pept Lett. 2021;28(12):1379-1390. doi: 10.2174/0929866528666210929163307. Protein Pept Lett. 2021. PMID: 34587878
-
Why less is more when generating tryptic peptides in bottom-up proteomics.Proteomics. 2014 Sep;14(17-18):2031-41. doi: 10.1002/pmic.201300479. Epub 2014 Aug 13. Proteomics. 2014. PMID: 25044798
-
Proteome-wide structural changes measured with limited proteolysis-mass spectrometry: an advanced protocol for high-throughput applications.Nat Protoc. 2023 Mar;18(3):659-682. doi: 10.1038/s41596-022-00771-x. Epub 2022 Dec 16. Nat Protoc. 2023. PMID: 36526727 Review.
-
Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics.Molecules. 2019 Feb 15;24(4):701. doi: 10.3390/molecules24040701. Molecules. 2019. PMID: 30781343 Free PMC article. Review.
Cited by
-
Protein Folding Stability Profiling of Colorectal Cancer Chemoresistance Identifies Functionally Relevant Biomarkers.J Proteome Res. 2023 Jun 2;22(6):1923-1935. doi: 10.1021/acs.jproteome.3c00045. Epub 2023 May 1. J Proteome Res. 2023. PMID: 37126456 Free PMC article.
-
Comparative Analysis of Protein Folding Stability-Based Profiling Methods for Characterization of Biological Phenotypes.J Am Soc Mass Spectrom. 2023 Mar 1;34(3):383-393. doi: 10.1021/jasms.2c00248. Epub 2023 Feb 20. J Am Soc Mass Spectrom. 2023. PMID: 36802530 Free PMC article.
-
Comparative Analysis of Mass-Spectrometry-Based Proteomic Methods for Protein Target Discovery Using a One-Pot Approach.J Am Soc Mass Spectrom. 2020 Feb 5;31(2):217-226. doi: 10.1021/jasms.9b00041. Epub 2019 Nov 22. J Am Soc Mass Spectrom. 2020. PMID: 32031398 Free PMC article.
-
FLiPPR: A Processor for Limited Proteolysis (LiP) Mass Spectrometry Datasets Built on FragPipe.bioRxiv [Preprint]. 2023 Dec 5:2023.12.04.569947. doi: 10.1101/2023.12.04.569947. bioRxiv. 2023. Update in: J Proteome Res. 2024 Jul 5;23(7):2332-2342. doi: 10.1021/acs.jproteome.3c00887. PMID: 38106106 Free PMC article. Updated. Preprint.
-
Mass-spectrometry-based proteomics: from single cells to clinical applications.Nature. 2025 Feb;638(8052):901-911. doi: 10.1038/s41586-025-08584-0. Epub 2025 Feb 26. Nature. 2025. PMID: 40011722 Review.
References
-
- Feng Y; De Franceschi G; Kahraman A; Soste M; Melnik A; Boersema PJ; de Laureto PP; Nikolaev Y; Oliveira AP; Picotti P, Global analysis of protein structural changes in complex proteomes. Nat Biotechnol 2014, 32 (10), 1036–1044. - PubMed
-
- Lomenick B; Hao R; Jonai N; Chin RM; Aghajan M; Warburton S; Wang J; Wu RP; Gomez F; Loo JA; Wohlschlegel JA; Vondriska TM; Pelletier J; Herschman HR; Clardy J; Clarke CF; Huang J, Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A 2009, 106 (51), 21984–21989. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
