

PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia
- PMID: 30404791
- PMCID: PMC6279404
- DOI: 10.1084/jem.20180570
PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia
Abstract
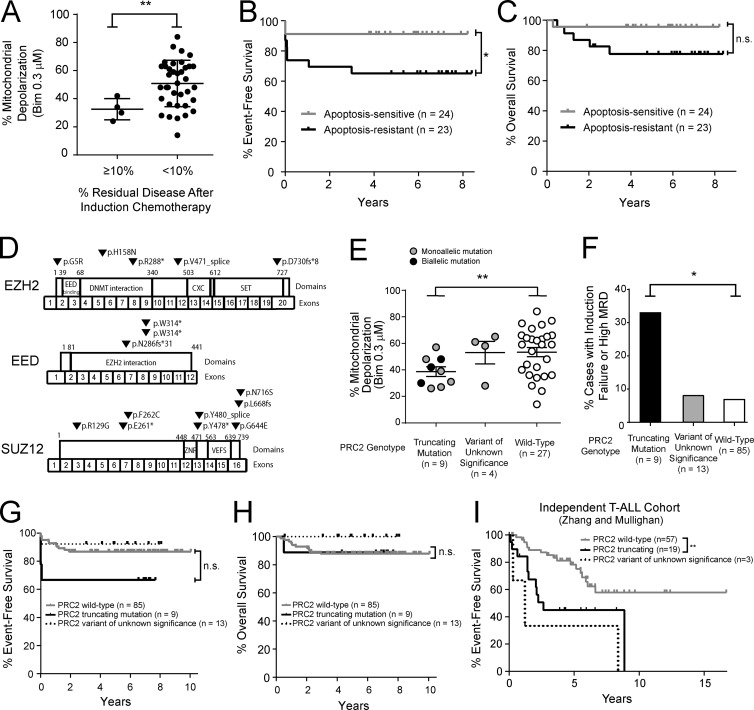
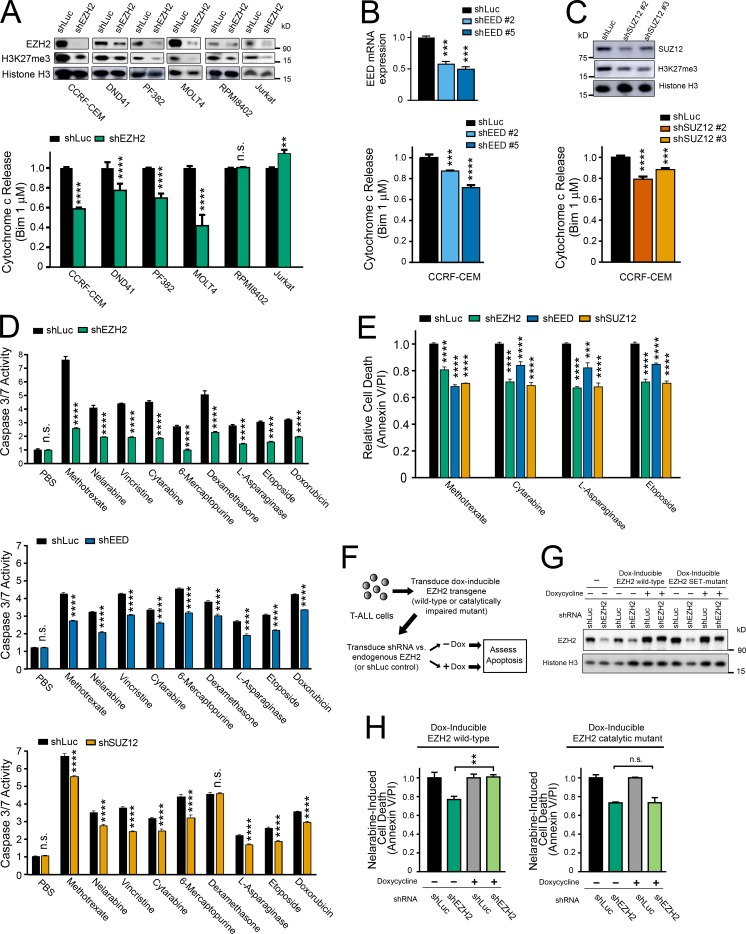
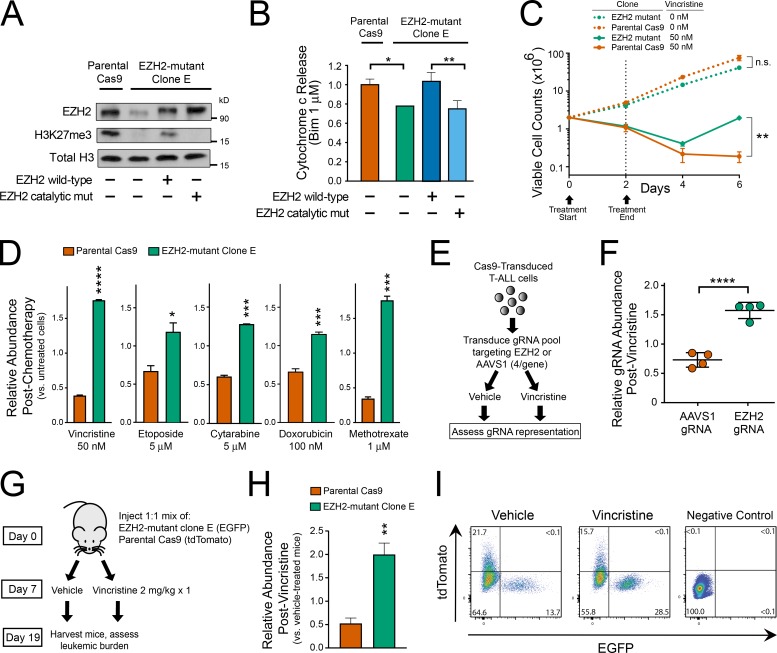
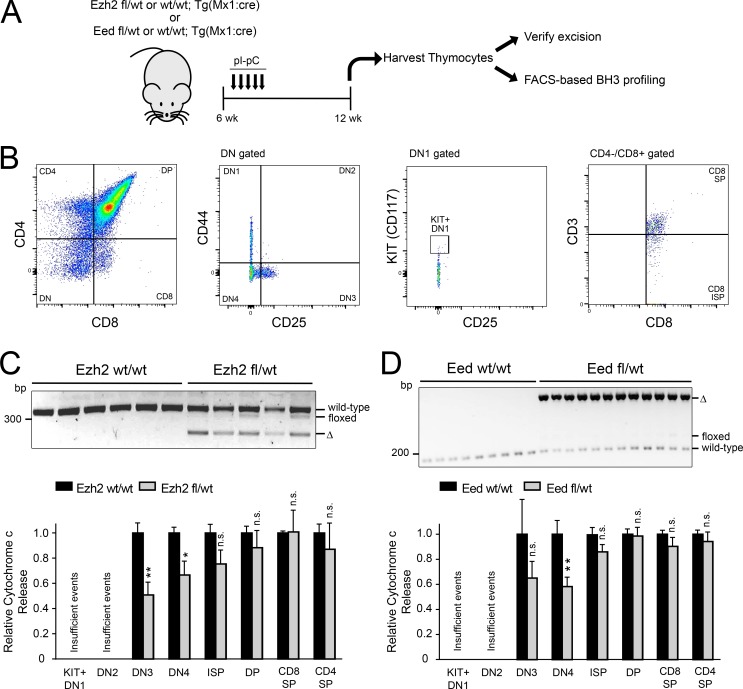
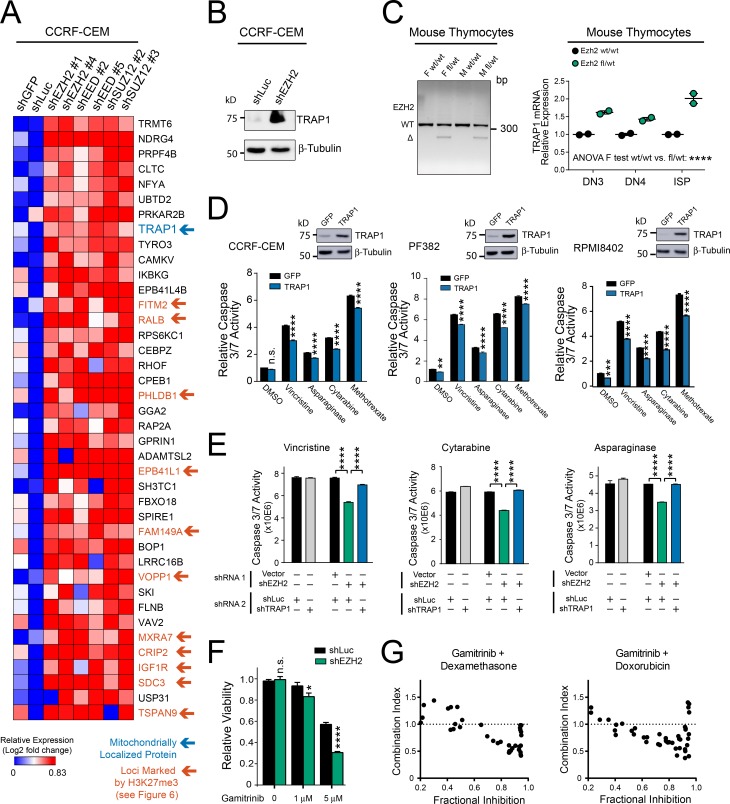
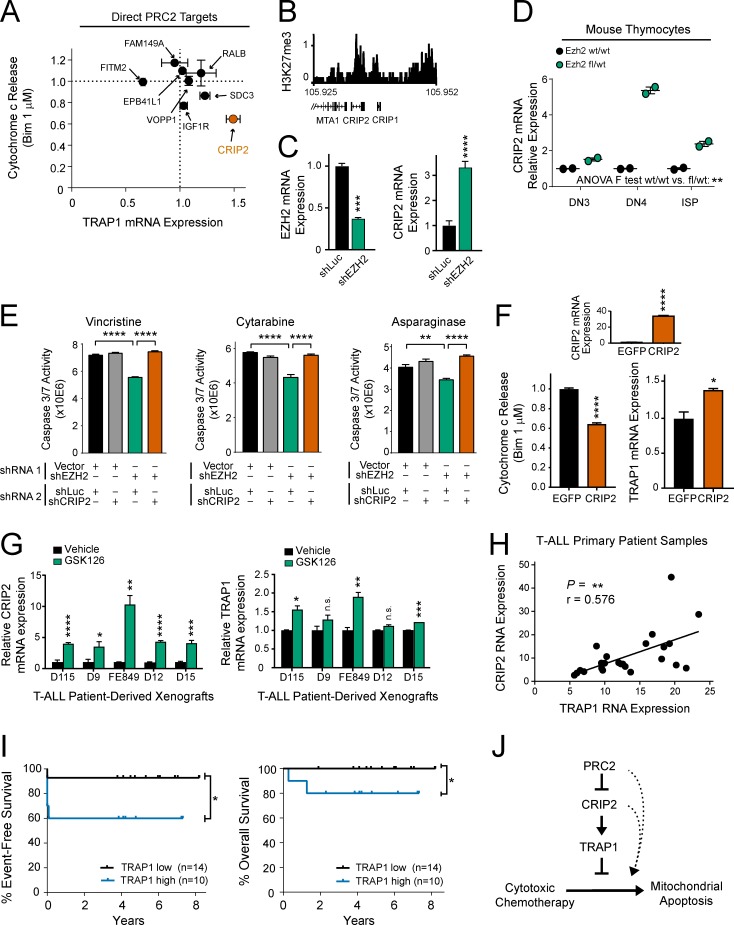
The tendency of mitochondria to undergo or resist BCL2-controlled apoptosis (so-called mitochondrial priming) is a powerful predictor of response to cytotoxic chemotherapy. Fully exploiting this finding will require unraveling the molecular genetics underlying phenotypic variability in mitochondrial priming. Here, we report that mitochondrial apoptosis resistance in T cell acute lymphoblastic leukemia (T-ALL) is mediated by inactivation of polycomb repressive complex 2 (PRC2). In T-ALL clinical specimens, loss-of-function mutations of PRC2 core components (EZH2, EED, or SUZ12) were associated with mitochondrial apoptosis resistance. In T-ALL cells, PRC2 depletion induced resistance to apoptosis induction by multiple chemotherapeutics with distinct mechanisms of action. PRC2 loss induced apoptosis resistance via transcriptional up-regulation of the LIM domain transcription factor CRIP2 and downstream up-regulation of the mitochondrial chaperone TRAP1 These findings demonstrate the importance of mitochondrial apoptotic priming as a prognostic factor in T-ALL and implicate mitochondrial chaperone function as a molecular determinant of chemotherapy response.
© 2018 Ariës et al.
Figures
References
-
- Abedalthagafi M.S., Merrill P.H., Bi W.L., Jones R.T., Listewnik M.L., Ramkissoon S.H., Thorner A.R., Dunn I.F., Beroukhim R., Alexander B.M., et al. 2014. Angiomatous meningiomas have a distinct genetic profile with multiple chromosomal polysomies including polysomy of chromosome 5. Oncotarget. 5:10596–10606. 10.18632/oncotarget.2517 - DOI - PMC - PubMed
-
- Baines C.P., Kaiser R.A., Purcell N.H., Blair N.S., Osinska H., Hambleton M.A., Brunskill E.W., Sayen M.R., Gottlieb R.A., Dorn G.W., et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 434:658–662. 10.1038/nature03434 - DOI - PubMed
-
- Bhola P.D., Mar B.G., Lindsley R.C., Ryan J.A., Hogdal L.J., Vo T.T., DeAngelo D.J., Galinsky I., Ebert B.L., and Letai A.. 2016. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J. Clin. Invest. 126:3827–3836. 10.1172/JCI82908 - DOI - PMC - PubMed
-
- Burns M.A., Liao Z.W., Yamagata N., Pouliot G.P., Stevenson K.E., Neuberg D.S., Thorner A.R., Ducar M., Silverman E.A., Hunger S.P., et al. 2018. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 32:2126–2137. 10.1038/s41375-018-0097-x - DOI - PMC - PubMed
-
- Cheung A.K., Ko J.M., Lung H.L., Chan K.W., Stanbridge E.J., Zabarovsky E., Tokino T., Kashima L., Suzuki T., Kwong D.L., et al. 2011. Cysteine-rich intestinal protein 2 (CRIP2) acts as a repressor of NF-kappaB-mediated proangiogenic cytokine transcription to suppress tumorigenesis and angiogenesis. Proc. Natl. Acad. Sci. USA. 108:8390–8395. 10.1073/pnas.1101747108 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
