

Herpes Simplex Virus 1 ICP22 Suppresses CD80 Expression by Murine Dendritic Cells
- PMID: 30404803
- PMCID: PMC6340034
- DOI: 10.1128/JVI.01803-18
Herpes Simplex Virus 1 ICP22 Suppresses CD80 Expression by Murine Dendritic Cells
Abstract
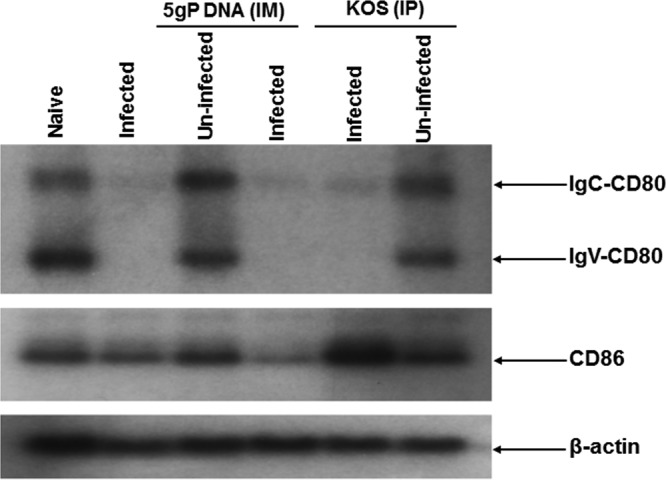
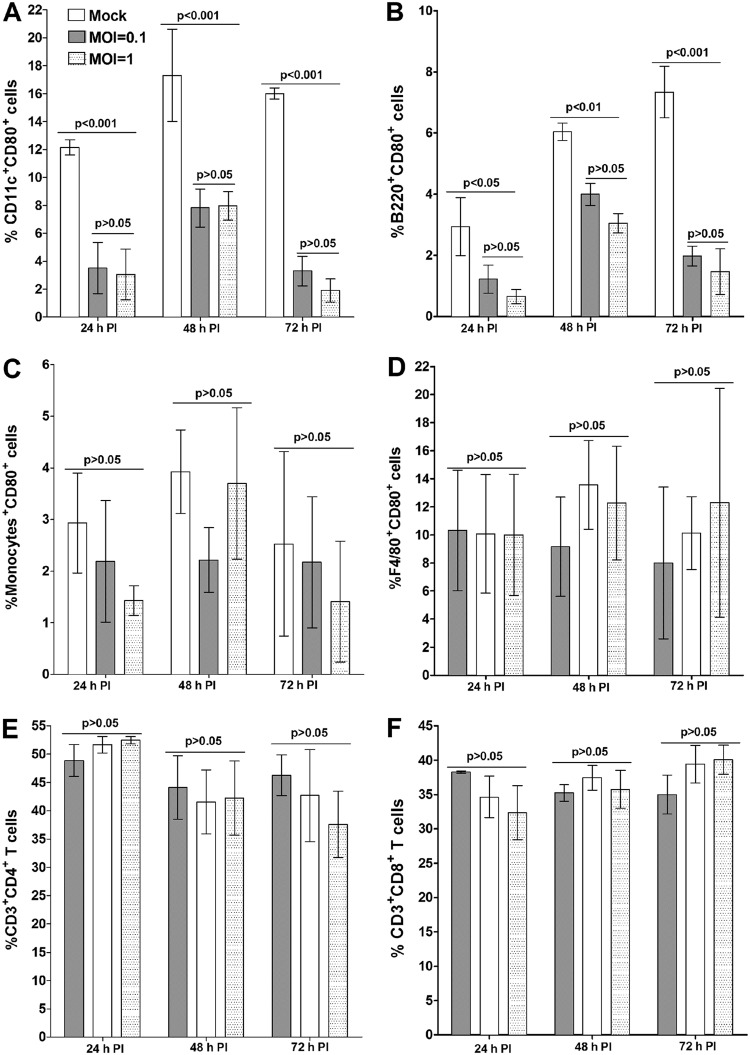
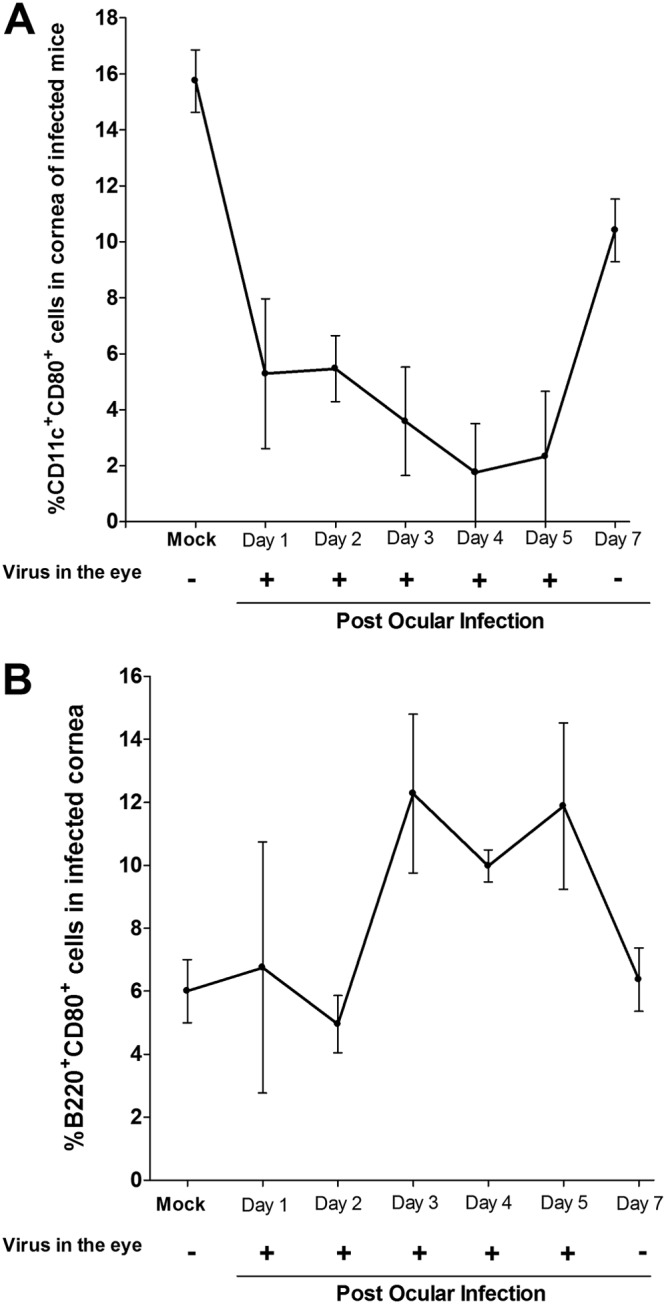
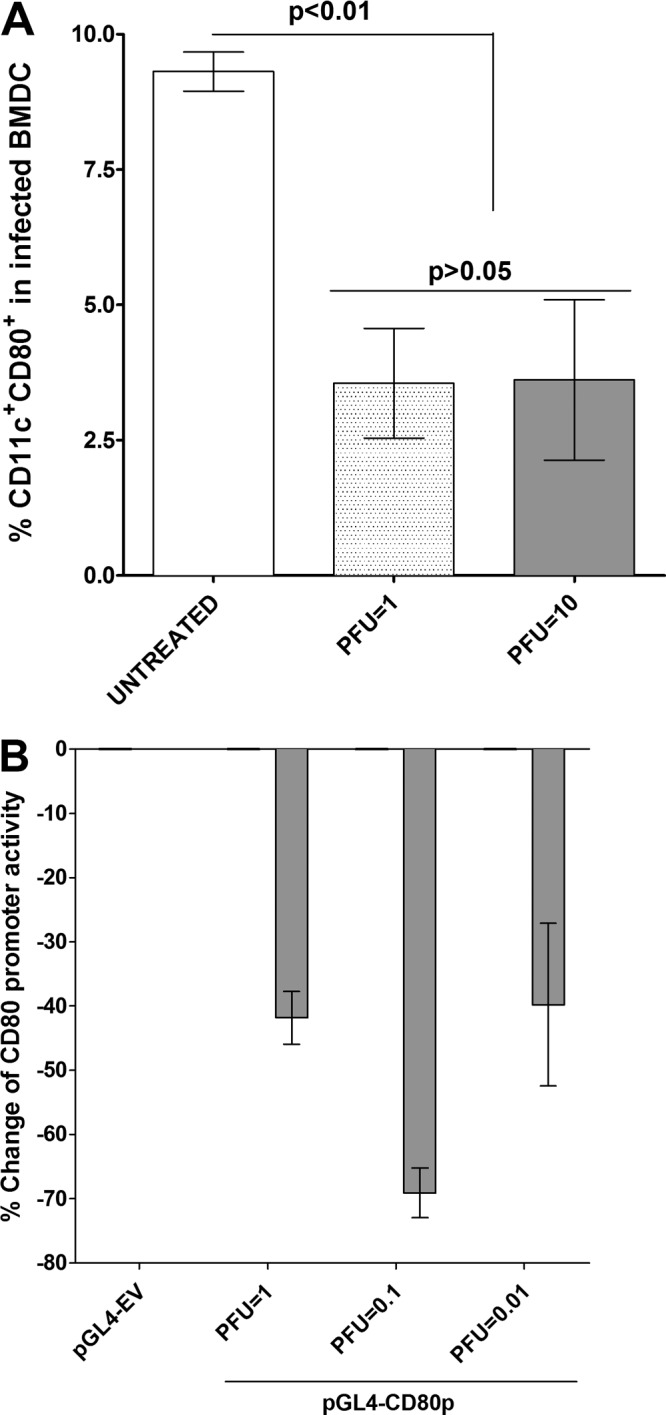
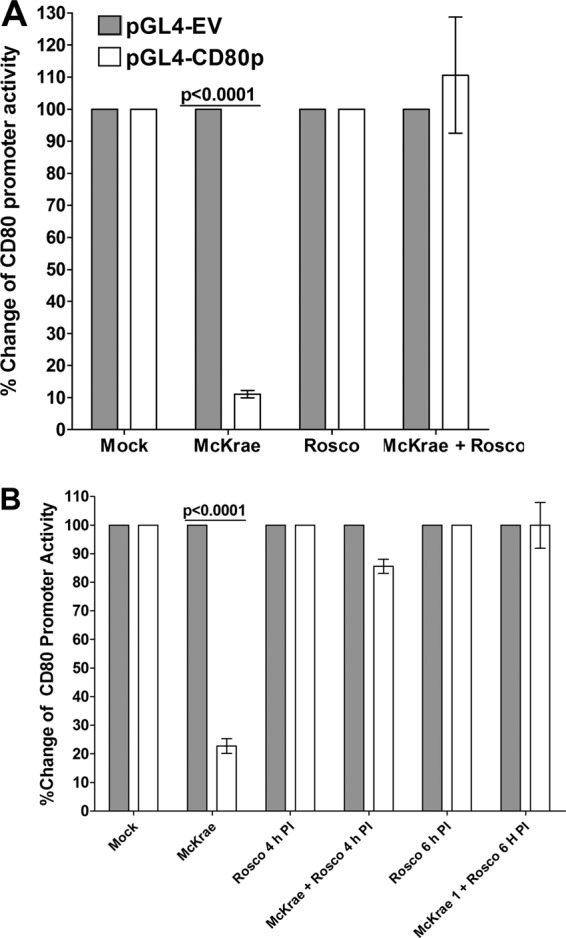
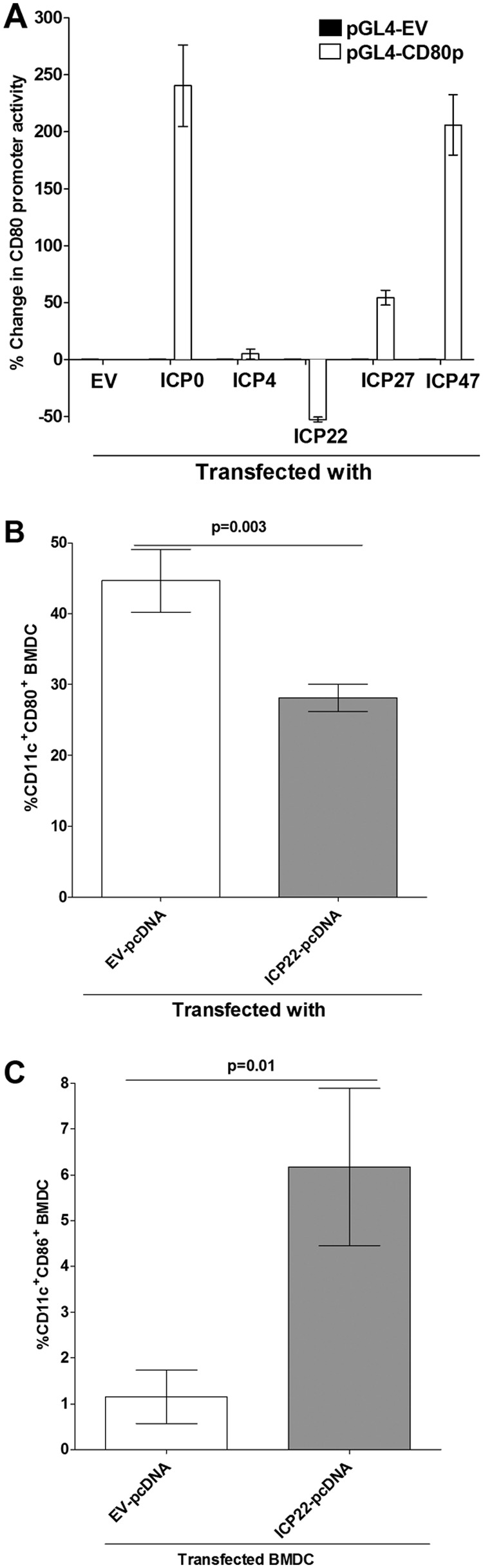
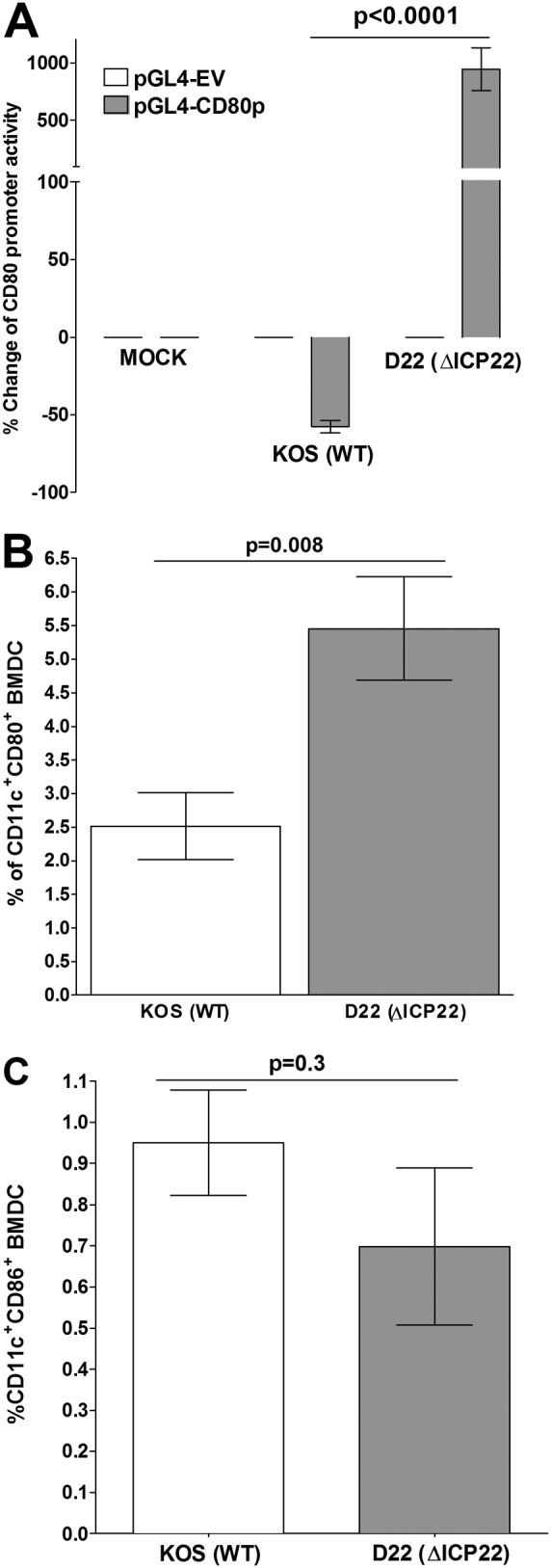
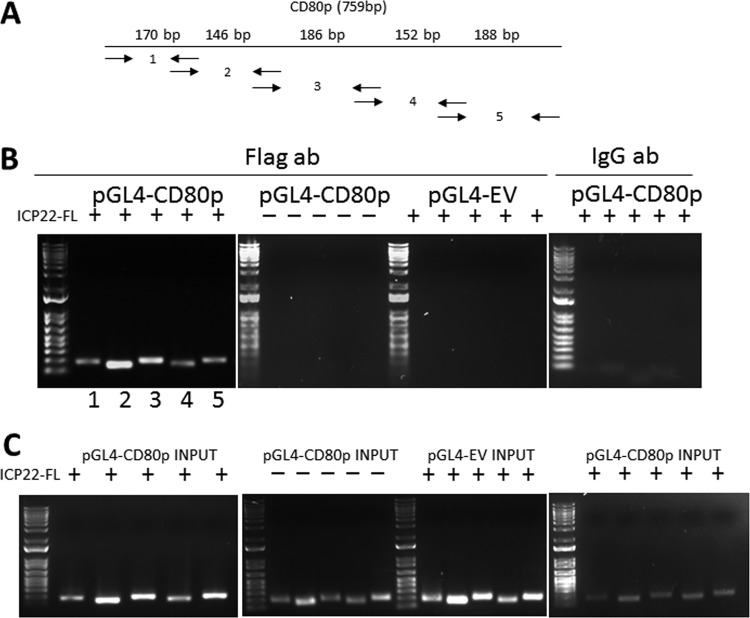
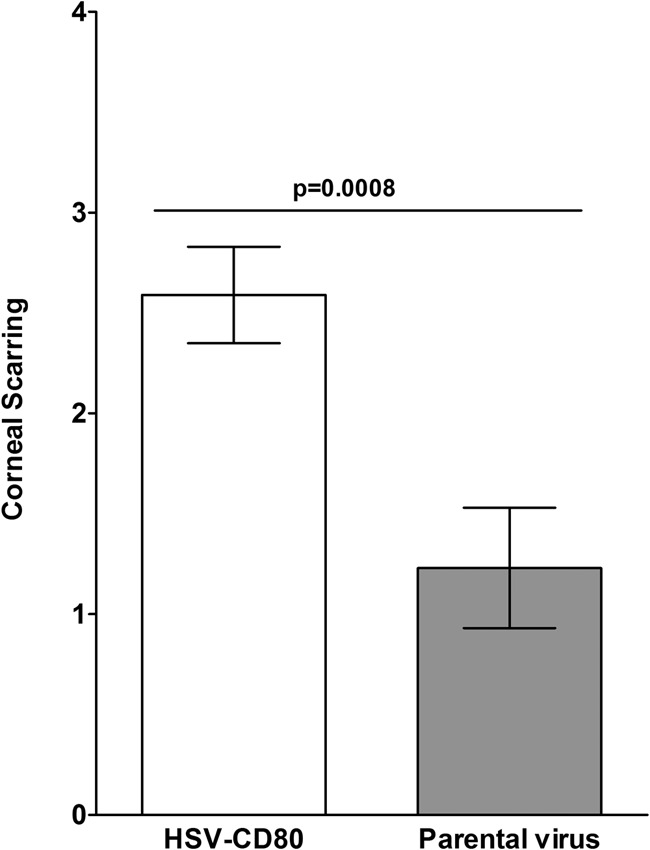
Herpes simplex virus type 1 (HSV-1) has the ability to delay its clearance from the eye during ocular infection. Here, we show that ocular infection of mice with HSV-1 suppressed expression of the costimulatory molecule CD80 but not CD86 in the cornea. The presence of neutralizing anti-HSV-1 antibodies did not alleviate this suppression. At the cellular level, HSV-1 consistently downregulated the expression of CD80 by dendritic cells (DCs) but not by other antigen-presenting cells. Furthermore, flow cytometric analysis of HSV-1-infected corneal cells during a 7-day period reduced CD80 expression in DCs but not in B cells, macrophages, or monocytes. This suppression was associated with the presence of virus. Similar results were obtained using infected or transfected spleen cells or bone marrow-derived DCs. A combination of roscovitine treatment, transfection with immediate early genes (IE), and infection with a recombinant HSV-1 lacking the ICP22 gene shows the importance of ICP22 in downregulation of the CD80 promoter but not the CD86 promoter in vitro and in vivo At the mechanistic level, we show that the HSV-1 immediate early gene ICP22 binds the CD80 promoter and that this interaction is required for HSV-1-mediated suppression of CD80 expression. Conversely, forced expression of CD80 by ocular infection of mice with a recombinant HSV-1 exacerbated corneal scarring in infected mice. Taken together, these studies identify ICP22-mediated suppression of CD80 expression in dendritic cells as central to delayed clearance of the virus and limitation of the cytopathological response to primary infection in the eye.IMPORTANCE HSV-1-induced eye disease is a major public health problem. Eye disease is associated closely with immune responses to the virus and is exacerbated by delayed clearance of the primary infection. The immune system relies on antigen-presenting cells of the innate immune system to activate the T cell response. We found that HSV-1 utilizes a robust and finely targeted mechanism of local immune evasion. It downregulates the expression of the costimulatory molecule CD80 but not CD86 on resident dendritic cells irrespective of the presence of anti-HSV-1 antibodies. The effect is mediated by direct binding of HSV-1 ICP22, the product of an immediate early gene of HSV-1, to the promoter of CD80. This immune evasion mechanism dampens the host immune response and, thus, reduces eye disease in ocularly infected mice. Therefore, ICP22 may be a novel inhibitor of CD80 that could be used to modulate the immune response.
Keywords: antigen-presenting cells; cornea; corneal scarring; eye disease; immediate early genes; immune suppression; virus replication.
Copyright © 2019 American Society for Microbiology.
Figures
References
-
- Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA, for the Herpetic Eye Disease Study Group. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882 doi:10.1016/S0161-6420(13)31155-5. - DOI - PubMed
-
- Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. doi:10.1136/bjo.80.11.969. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
