

In silico Prediction, Characterization, Molecular Docking, and Dynamic Studies on Fungal SDRs as Novel Targets for Searching Potential Fungicides Against Fusarium Wilt in Tomato
- PMID: 30405403
- PMCID: PMC6204350
- DOI: 10.3389/fphar.2018.01038
In silico Prediction, Characterization, Molecular Docking, and Dynamic Studies on Fungal SDRs as Novel Targets for Searching Potential Fungicides Against Fusarium Wilt in Tomato
Abstract
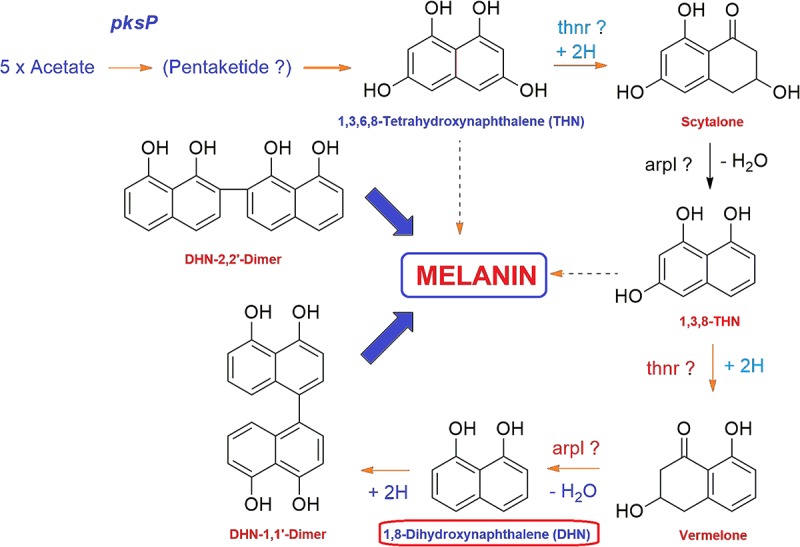
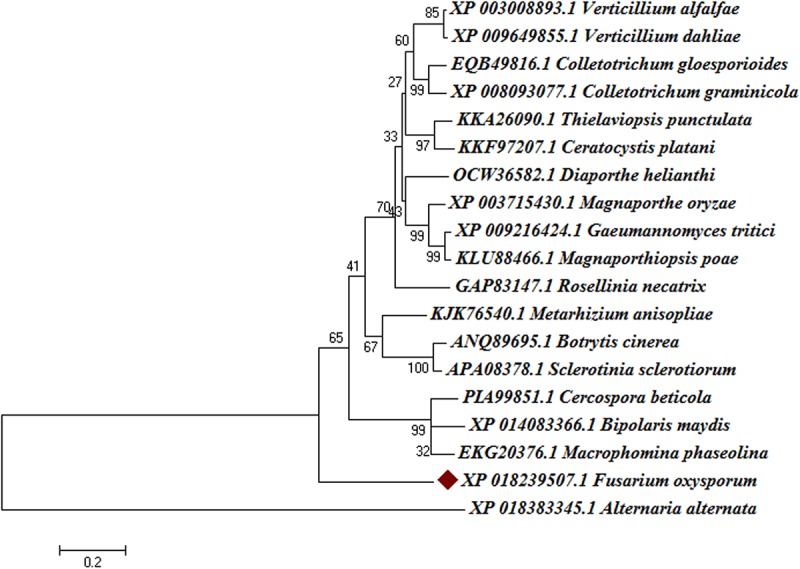
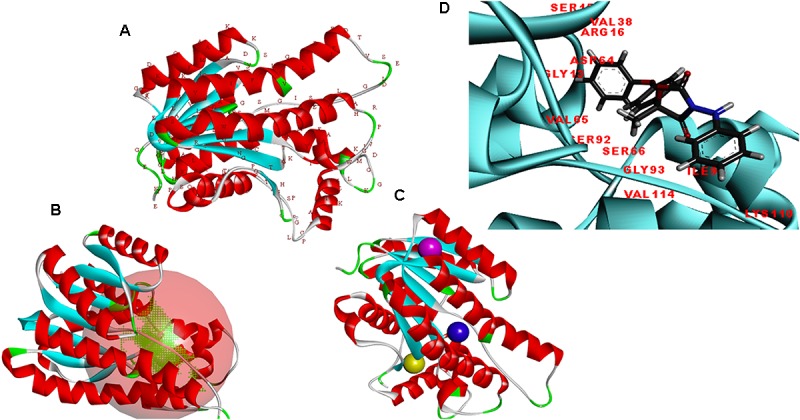
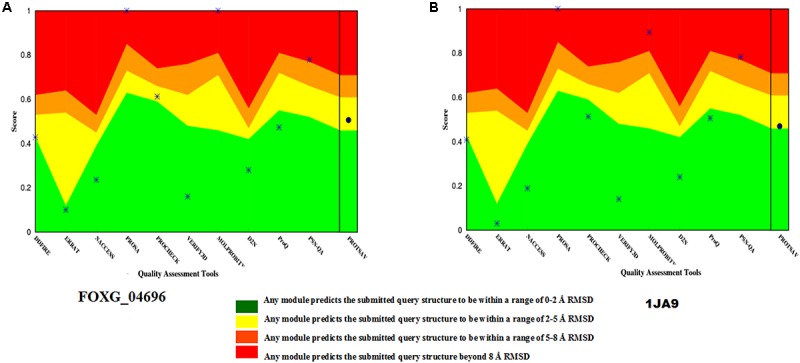
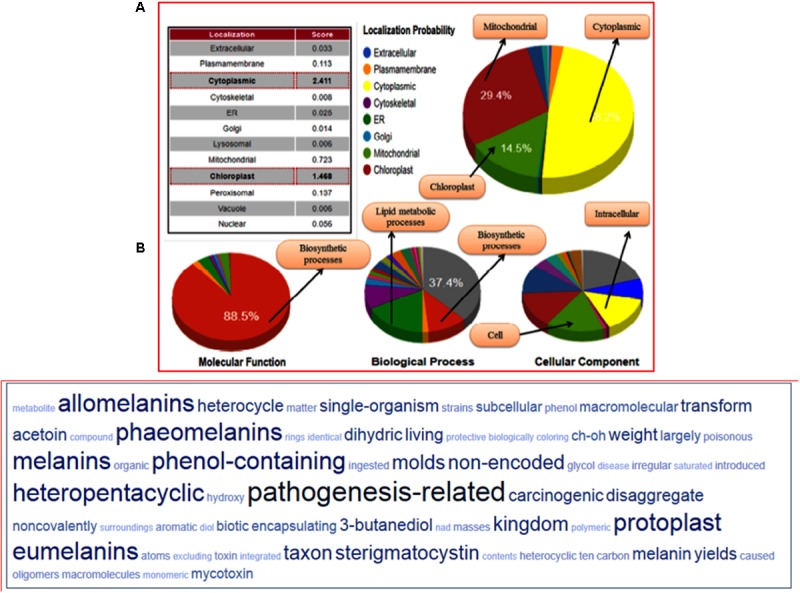
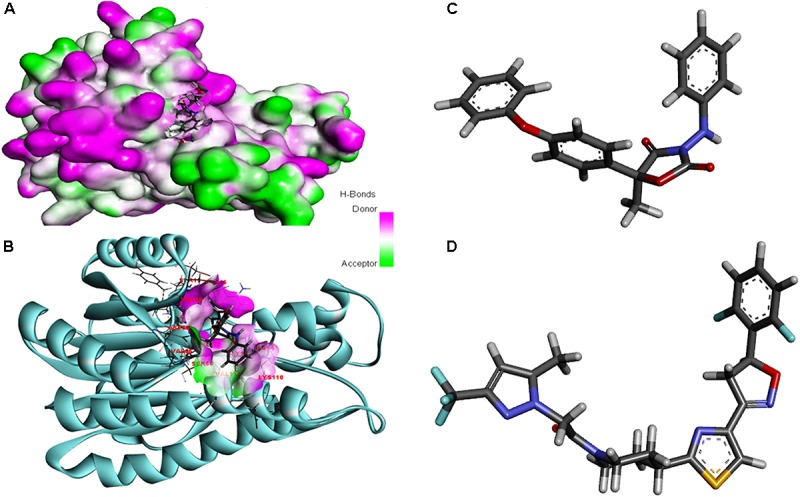
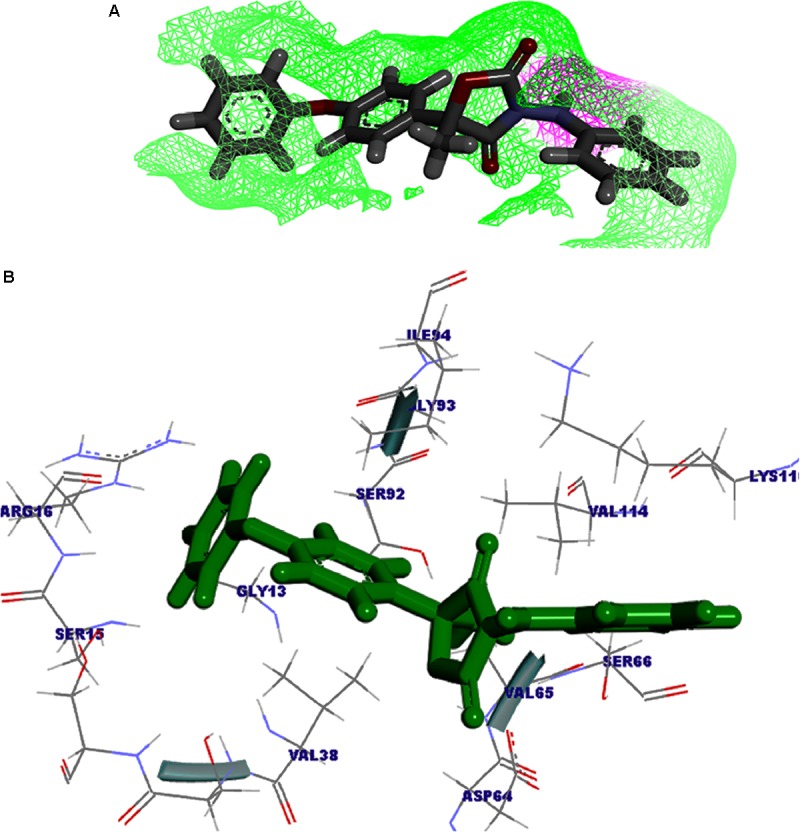
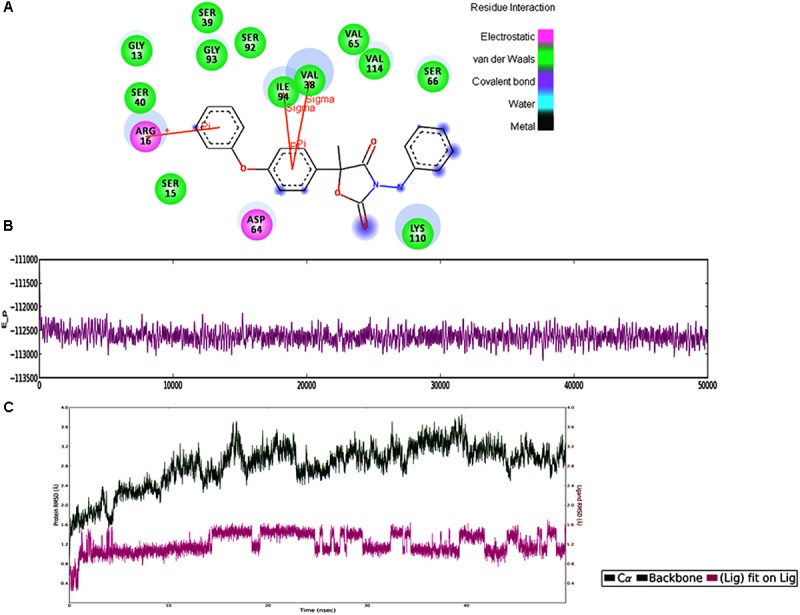
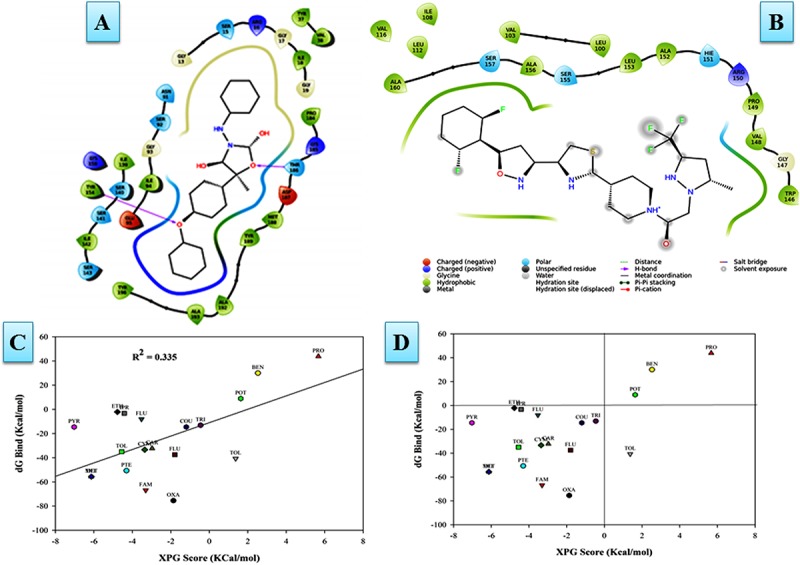
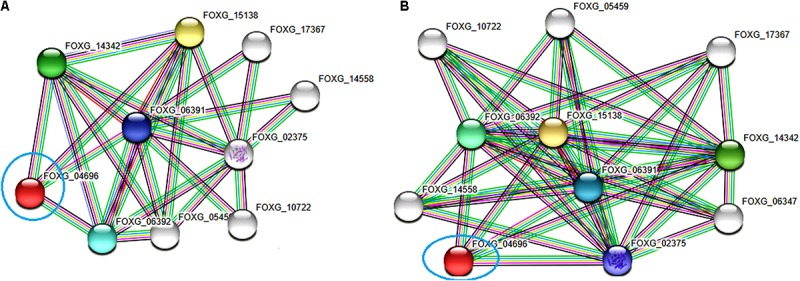
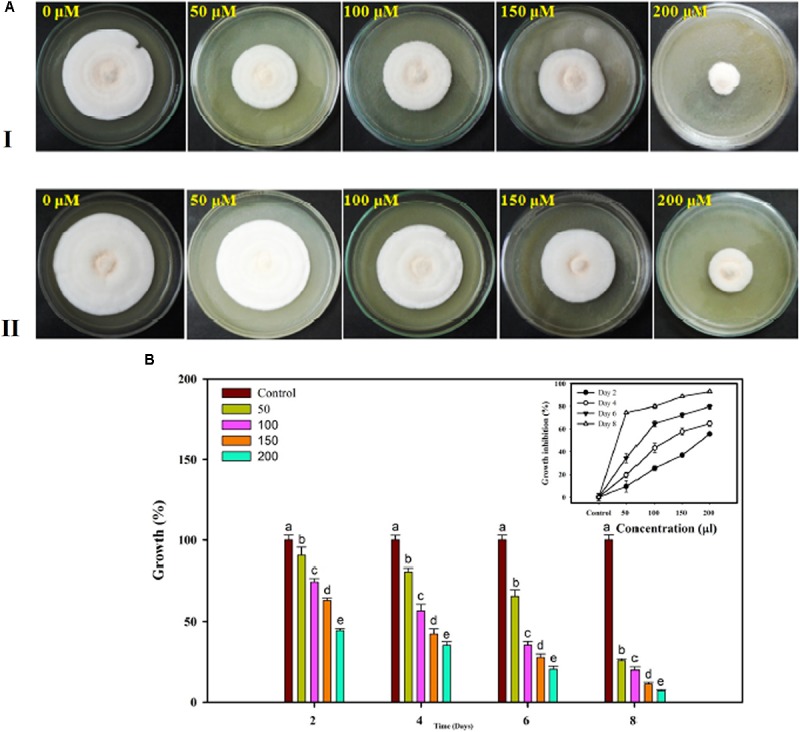
Vascular wilt of tomato caused by Fusarium oxysporum f.sp. lycopersici (FOL) is one of the most devastating diseases, that delimits the tomato production worldwide. Fungal short-chain dehydrogenases/reductases (SDRs) are NADP(H) dependent oxidoreductases, having shared motifs and common functional mechanism, have been demonstrated as biochemical targets for commercial fungicides. The 1,3,6,8 tetra hydroxynaphthalene reductase (T4HNR) protein, a member of SDRs family, catalyzes the naphthol reduction reaction in fungal melanin biosynthesis. We retrieved an orthologous member of T4HNR, (complexed with NADP(H) and pyroquilon from Magnaporthe grisea) in the FOL (namely; FOXG_04696) based on homology search, percent identity and sequence similarity (93% query cover; 49% identity). The hypothetical protein FOXG_04696 (T4HNR like) had conserved T-G-X-X-X-G-X-G motif (cofactor binding site) at N-terminus, similar to M. grisea (1JA9) and Y-X-X-X-K motif, as a part of the active site, bearing homologies with two fungal keto reductases T4HNR (M. grisea) and 17-β-hydroxysteroid dehydrogenase from Curvularia lunata (teleomorph: Cochliobolus lunatus PDB ID: 3IS3). The catalytic tetrad of T4HNR was replaced with ASN115, SER141, TYR154, and LYS158 in the FOXG_04696. The structural alignment and superposition of FOXG_04696 over the template proteins (3IS3 and 1JA9) revealed minimum RMSD deviations of the C alpha atomic coordinates, and therefore, had structural conservation. The best protein model (FOXG_04696) was docked with 37 fungicides, to evaluate their binding affinities. The Glide XP and YASARA docked complexes showed discrepancies in results, for scoring and ranking the binding affinities of fungicides. The docked complexes were further refined and rescored from their docked poses through 50 ns long MD simulations, and binding free energies (ΔGbind) calculations, using MM/GBSA analysis, revealed Oxathiapiprolin and Famoxadone as better fungicides among the selected one. However, Famoxadone had better interaction of the docked residues, with best protein ligand contacts, minimum RMSD (high accuracy of the docking pose) and RMSF (structural integrity and conformational flexibility of docking) at the specified docking site. The Famoxadone was found to be acceptable based on in silico toxicity and in vitro growth inhibition assessment. We conclude that the FOXG_04696, could be employed as a novel candidate protein, for structure-based design, and screening of target fungicides against the FOL pathogen.
Keywords: MD simulations; MM/GBSA analysis; THN reductase; fungicide; homology modeling; melanin; protein–fungicide interaction.
Figures

References
-
- Aamir M., Singh V. K., Meena M., Upadhyay R. S., Gupta V. K., Singh S. (2017). Structural and functional insights into WRKY3 and WRKY4 transcription factors to unravel the WRKY–DNA (W-Box) complex interaction in tomato (Solanum lycopersicum L.). A computational approach. Front. Plant Sci. 8:819. 10.3389/fpls.2017.00819 - DOI - PMC - PubMed
-
- Aamir M., Singh V. K., Dubey M. K., Kashyap S. P., Zehra A., Upadhyay R. S., et al. (2018). Structural and functional dissection of differentially expressed tomato WRKY transcripts in host defense response against the vascular wilt pathogen (Fusarium oxysporum f. sp. lycopersici). PLoS One 13:e0193922. 10.1371/journal.pone.0193922 - DOI - PMC - PubMed
-
- Abo Ellil A. H., Sharaf E. F. (2000). “Growth, morphological alteration and adaptation of some plant pathogenic fungi to benlate and dicarboximide; a new look,” in Proceedings of the 1st International Conference of Biological Sciences – Faculty of Science, Vol. 1 (Tanta: Tanta University; ), 568–579.
LinkOut - more resources
Full Text Sources
Miscellaneous