

Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches
- PMID: 30406384
- PMCID: PMC6277304
- DOI: 10.1007/s13311-018-00677-1
Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches
Abstract
Ryanodine receptor type 1-related myopathies (RYR1-RM) are the most common class of congenital myopathies. Historically, RYR1-RM classification and diagnosis have been guided by histopathologic findings on muscle biopsy. Main histological subtypes of RYR1-RM include central core disease, multiminicore disease, core-rod myopathy, centronuclear myopathy, and congenital fiber-type disproportion. A range of RYR1-RM clinical phenotypes has also emerged more recently and includes King Denborough syndrome, RYR1 rhabdomyolysis-myalgia syndrome, atypical periodic paralysis, congenital neuromuscular disease with uniform type 1 fibers, and late-onset axial myopathy. This expansion of the RYR1-RM disease spectrum is due, in part, to implementation of next-generation sequencing methods, which include the entire RYR1 coding sequence rather than being restricted to hotspot regions. These methods enhance diagnostic capabilities, especially given historic limitations of histopathologic and clinical overlap across RYR1-RM. Both dominant and recessive modes of inheritance have been documented, with the latter typically associated with a more severe clinical phenotype. As with all congenital myopathies, no FDA-approved treatments exist to date. Here, we review histopathologic, clinical, imaging, and genetic diagnostic features of the main RYR1-RM subtypes. We also discuss the current state of treatments and focus on disease-modulating (nongenetic) therapeutic strategies under development for RYR1-RM. Finally, perspectives for future approaches to treatment development are broached.
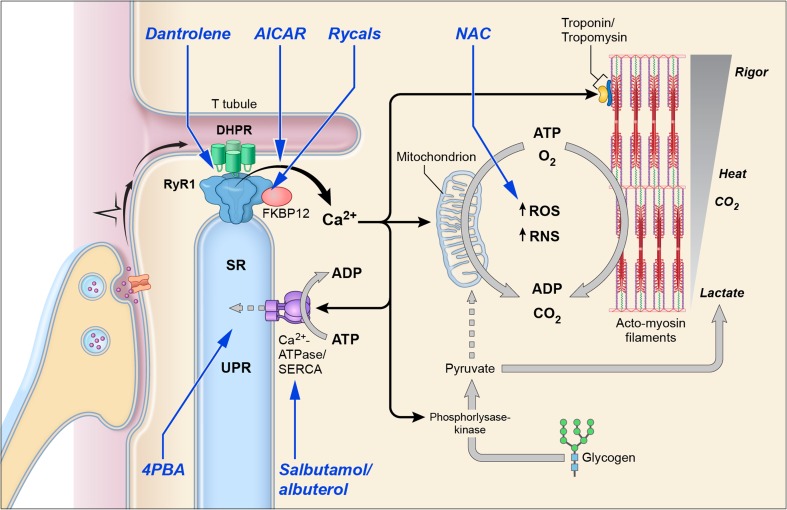
Keywords: 4PBA; Central core disease; Myopathies; N-acetylcysteine; RYR1; Rycal; Salbutamol; Therapeutics.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Dowling JJ, North KN, Goebel HH, Beggs AH. Chapter 28 - Congenital and Other Structural Myopathies A2 - Darras, Basil T. In: Jones HR, Ryan MM, Vivo DCD, editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Second Edition. San Diego: Academic Press; 2015. pp. 499–537.
-
- Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscular disorders : NMD. 2000;10(1):1–9. - PubMed
-
- Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ. Prevalence of congenital myopathies in a representative pediatric united states population. Annals of neurology. 2011;70(4):662–5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
