

Loss of CXCL12/CXCR4 signalling impacts several aspects of cardiovascular development but does not exacerbate Tbx1 haploinsufficiency
- PMID: 30408103
- PMCID: PMC6224166
- DOI: 10.1371/journal.pone.0207251
Loss of CXCL12/CXCR4 signalling impacts several aspects of cardiovascular development but does not exacerbate Tbx1 haploinsufficiency
Abstract
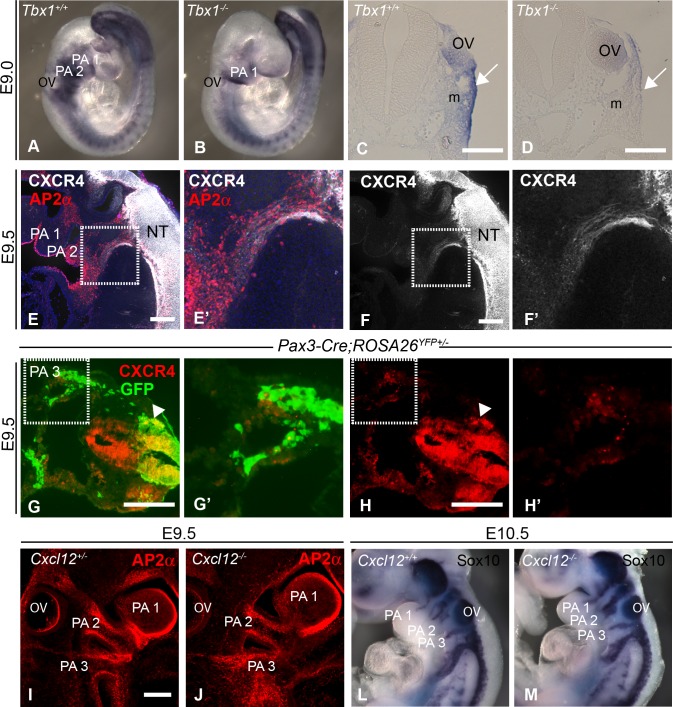
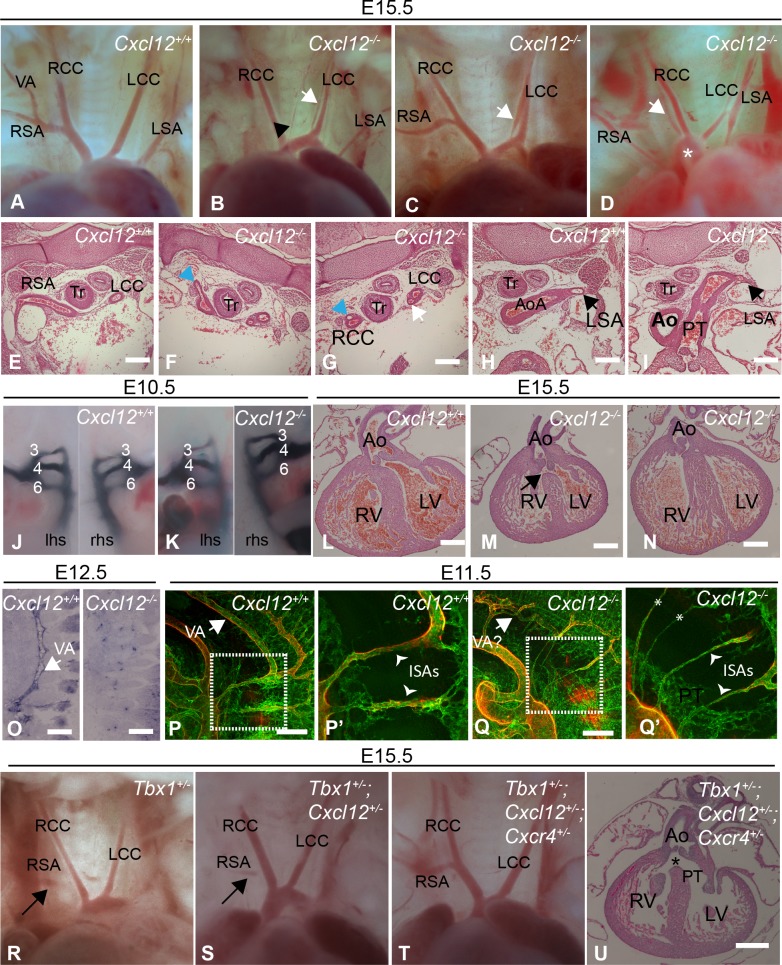
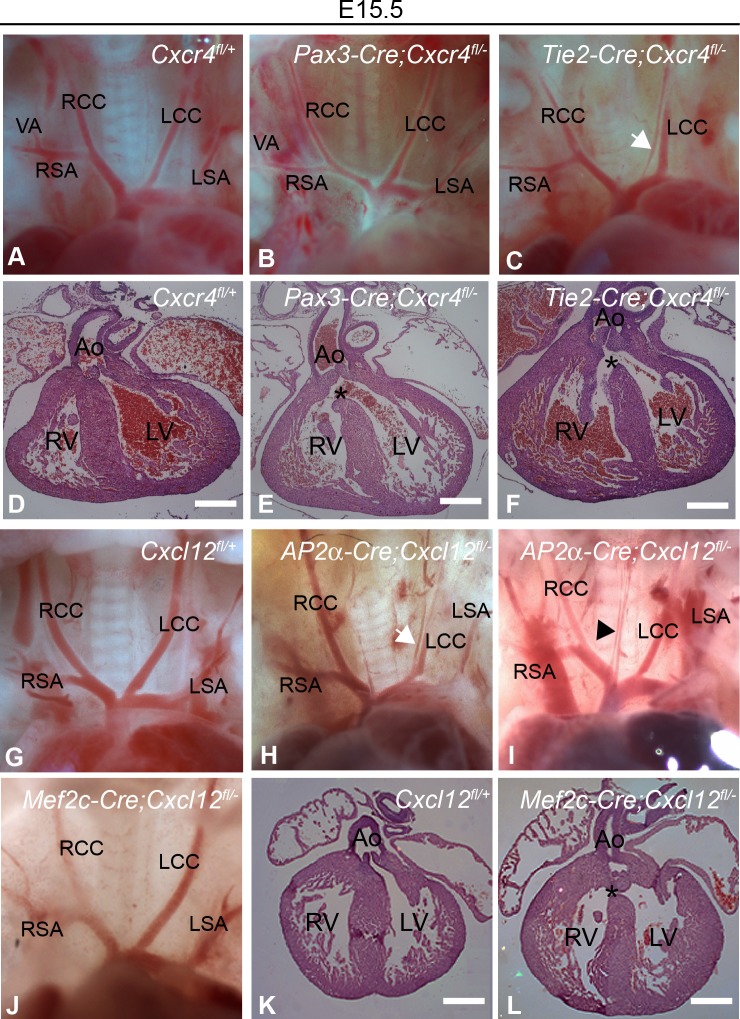
The CXCL12-CXCR4 pathway has crucial roles in stem cell homing and maintenance, neuronal guidance, cancer progression, inflammation, remote-conditioning, cell migration and development. Recently, work in chick suggested that signalling via CXCR4 in neural crest cells (NCCs) has a role in the 22q11.2 deletion syndrome (22q11.2DS), a disorder where haploinsufficiency of the transcription factor TBX1 is responsible for the major structural defects. We tested this idea in mouse models. Our analysis of genes with altered expression in Tbx1 mutant mouse models showed down-regulation of Cxcl12 in pharyngeal surface ectoderm and rostral mesoderm, both tissues with the potential to signal to migrating NCCs. Conditional mutagenesis of Tbx1 in the pharyngeal surface ectoderm is associated with hypo/aplasia of the 4th pharyngeal arch artery (PAA) and interruption of the aortic arch type B (IAA-B), the cardiovascular defect most typical of 22q11.2DS. We therefore analysed constitutive mouse mutants of the ligand (CXCL12) and receptor (CXCR4) components of the pathway, in addition to ectodermal conditionals of Cxcl12 and NCC conditionals of Cxcr4. However, none of these typical 22q11.2DS features were detected in constitutively or conditionally mutant embryos. Instead, duplicated carotid arteries were observed, a phenotype recapitulated in Tie-2Cre (endothelial) conditional knock outs of Cxcr4. Previous studies have demonstrated genetic interaction between signalling pathways and Tbx1 haploinsufficiency e.g. FGF, WNT, SMAD-dependent. We therefore tested for possible epistasis between Tbx1 and the CXCL12 signalling axis by examining Tbx1 and Cxcl12 double heterozygotes as well as Tbx1/Cxcl12/Cxcr4 triple heterozygotes, but failed to identify any exacerbation of the Tbx1 haploinsufficient arch artery phenotype. We conclude that CXCL12 signalling via NCC/CXCR4 has no major role in the genesis of the Tbx1 loss of function phenotype. Instead, the pathway has a distinct effect on remodelling of head vessels and interventricular septation mediated via CXCL12 signalling from the pharyngeal surface ectoderm and second heart field to endothelial cells.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
CXCL12-CXCR4 signalling plays an essential role in proper patterning of aortic arch and pulmonary arteries.Cardiovasc Res. 2017 Nov 1;113(13):1677-1687. doi: 10.1093/cvr/cvx188. Cardiovasc Res. 2017. PMID: 29016745 Free PMC article.
-
Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome.Circ Res. 2010 Mar 5;106(4):686-94. doi: 10.1161/CIRCRESAHA.109.205732. Epub 2010 Jan 28. Circ Res. 2010. PMID: 20110535 Free PMC article.
-
Reduced dosage of β-catenin provides significant rescue of cardiac outflow tract anomalies in a Tbx1 conditional null mouse model of 22q11.2 deletion syndrome.PLoS Genet. 2017 Mar 27;13(3):e1006687. doi: 10.1371/journal.pgen.1006687. eCollection 2017 Mar. PLoS Genet. 2017. PMID: 28346476 Free PMC article.
-
22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development.Pediatr Cardiol. 2010 Apr;31(3):378-90. doi: 10.1007/s00246-009-9613-0. Pediatr Cardiol. 2010. PMID: 20054531 Review.
-
Understanding the role of Tbx1 as a candidate gene for 22q11.2 deletion syndrome.Curr Allergy Asthma Rep. 2013 Dec;13(6):613-21. doi: 10.1007/s11882-013-0384-6. Curr Allergy Asthma Rep. 2013. PMID: 23996541 Free PMC article. Review.
Cited by
-
Key Regulatory Differentially Expressed Genes in the Blood of Atrial Septal Defect Children Treated With Occlusion Devices.Front Genet. 2021 Dec 8;12:790426. doi: 10.3389/fgene.2021.790426. eCollection 2021. Front Genet. 2021. PMID: 34956331 Free PMC article.
-
Dual role for CXCL12 signaling in semilunar valve development.Cell Rep. 2021 Aug 24;36(8):109610. doi: 10.1016/j.celrep.2021.109610. Cell Rep. 2021. PMID: 34433040 Free PMC article.
-
cxcl12a plays an essential role in pharyngeal cartilage development.Front Cell Dev Biol. 2023 Oct 4;11:1243265. doi: 10.3389/fcell.2023.1243265. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37860819 Free PMC article.
-
Involvement of CXCR4 in Normal and Abnormal Development.Cells. 2019 Feb 20;8(2):185. doi: 10.3390/cells8020185. Cells. 2019. PMID: 30791675 Free PMC article. Review.
-
Associations between birth defects and childhood and adolescent germ cell tumors according to sex, histologic subtype, and site.Cancer. 2023 Oct 15;129(20):3300-3308. doi: 10.1002/cncr.34906. Epub 2023 Jun 27. Cancer. 2023. PMID: 37366624 Free PMC article.
References
-
- Lewin MB, Lindsay EA, Jurecic V, Goytia V, Towbin JA, Baldini A. A genetic etiology for interruption of the aortic arch type B. Am J Cardiol. 1997;80(4):493–7. - PubMed
-
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;11(8):915–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
