

How quantitative is metabarcoding: A meta-analytical approach
- PMID: 30408260
- PMCID: PMC7379500
- DOI: 10.1111/mec.14920
How quantitative is metabarcoding: A meta-analytical approach
Abstract
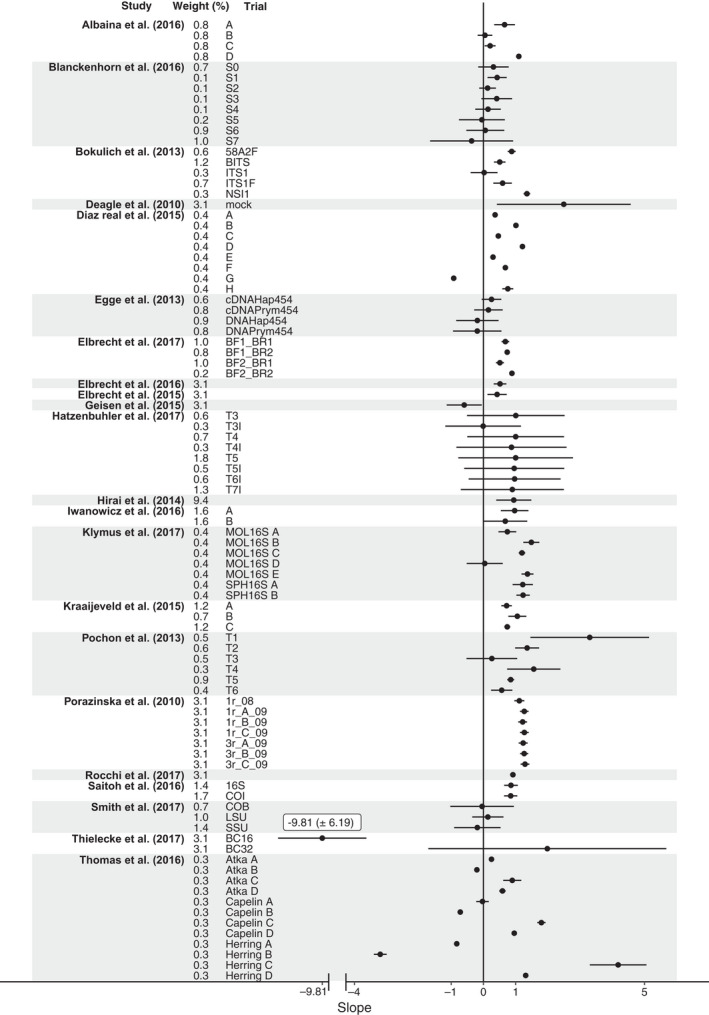
Metabarcoding has been used in a range of ecological applications such as taxonomic assignment, dietary analysis and the analysis of environmental DNA. However, after a decade of use in these applications there is little consensus on the extent to which proportions of reads generated corresponds to the original proportions of species in a community. To quantify our current understanding, we conducted a structured review and meta-analysis. The analysis suggests that a weak quantitative relationship may exist between the biomass and sequences produced (slope = 0.52 ± 0.34, p < 0.01), albeit with a large degree of uncertainty. None of the tested moderators, sequencing platform type, the number of species used in a trial or the source of DNA, were able to explain the variance. Our current understanding of the factors affecting the quantitative performance of metabarcoding is still limited: additional research is required before metabarcoding can be confidently utilized for quantitative applications. Until then, we advocate the inclusion of mock communities when metabarcoding as this facilitates direct assessment of the quantitative ability of any given study.
Keywords: biomass; high-throughput sequencing; meta-analysis; metabarcoding; next-generation sequencing.
© 2018 The Authors. Molecular Ecology Published by John Wiley & Sons Ltd.
Figures
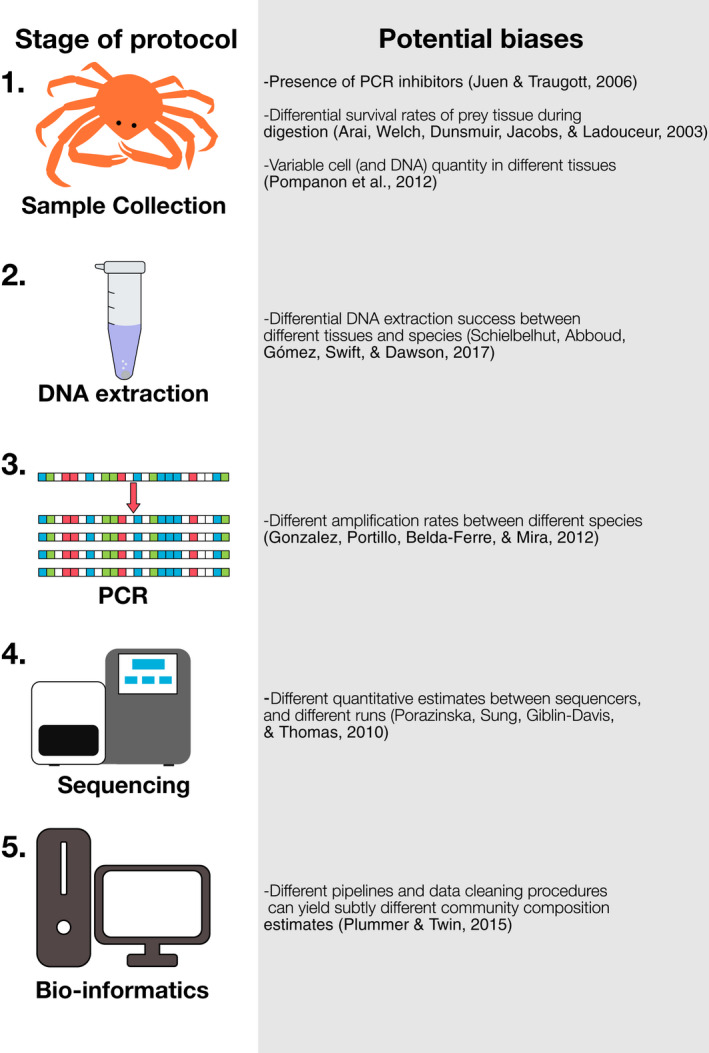
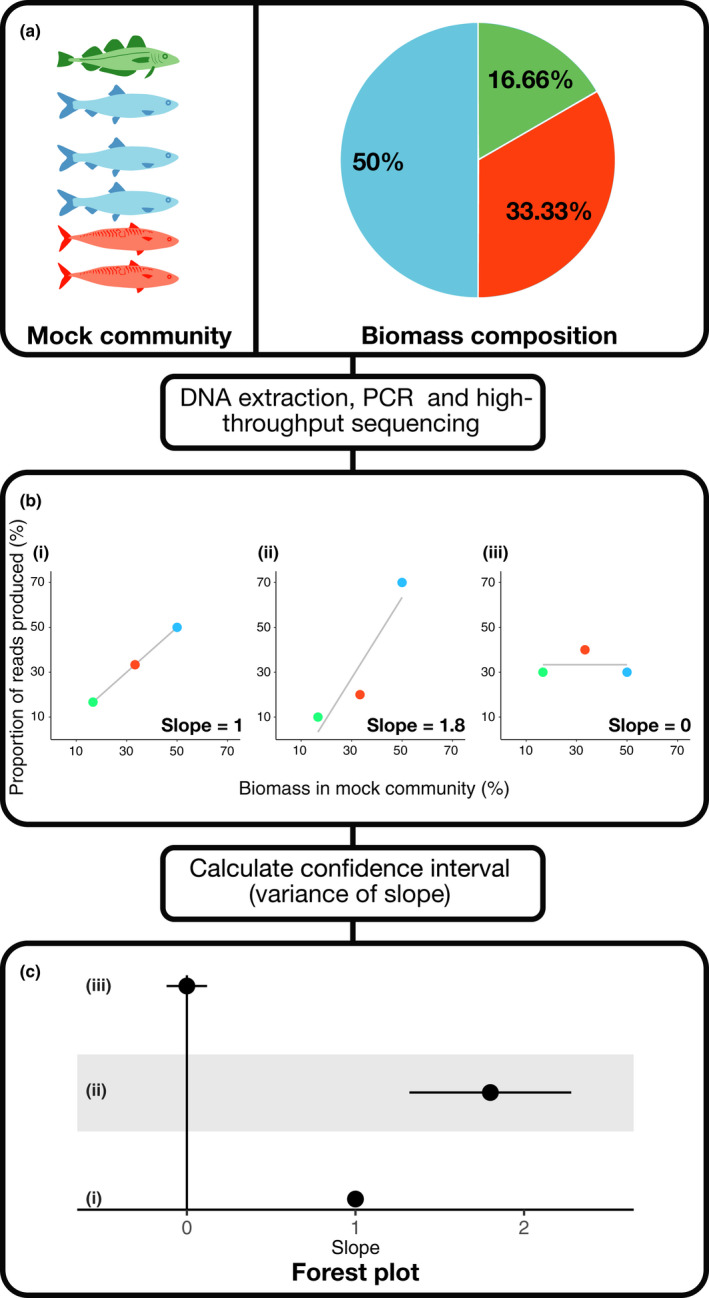
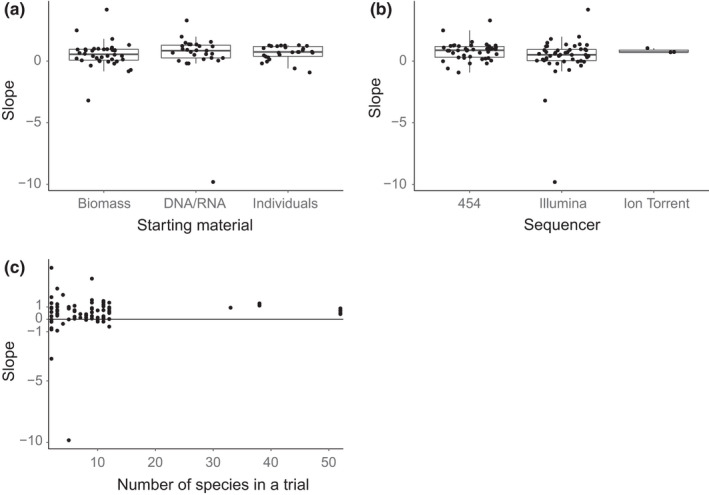
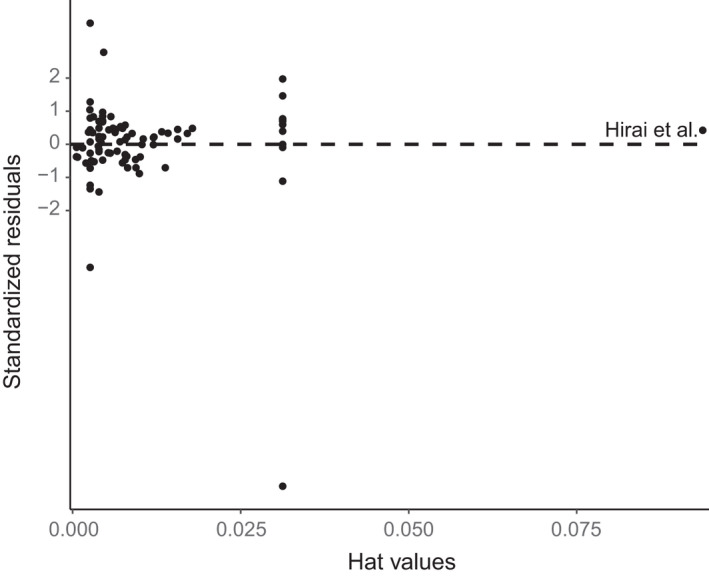
References
-
- Arai, M. N. , Welch, D. W. , Dunsmuir, A. L. , Jacobs, M. C. , & Ladouceur, A. R. (2003). Digestion of pelagic Ctenophora and Cnidaria by fish. Canadian Journal of Fisheries and Aquatic Sciences, 60, 825–829. 10.1139/f03-071 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
