

Visualization-assisted binning of metagenome assemblies reveals potential new pathogenic profiles in idiopathic travelers' diarrhea
- PMID: 30409177
- PMCID: PMC6225641
- DOI: 10.1186/s40168-018-0579-0
Visualization-assisted binning of metagenome assemblies reveals potential new pathogenic profiles in idiopathic travelers' diarrhea
Abstract
Background: Travelers' diarrhea (TD) is often caused by enterotoxigenic Escherichia coli, enteroaggregative E. coli, other bacterial pathogens, Norovirus, and occasionally parasites. Nevertheless, standard diagnostic methods fail to identify pathogens in more than 40% of TD patients. It is predicted that new pathogens may be causative agents of the disease.
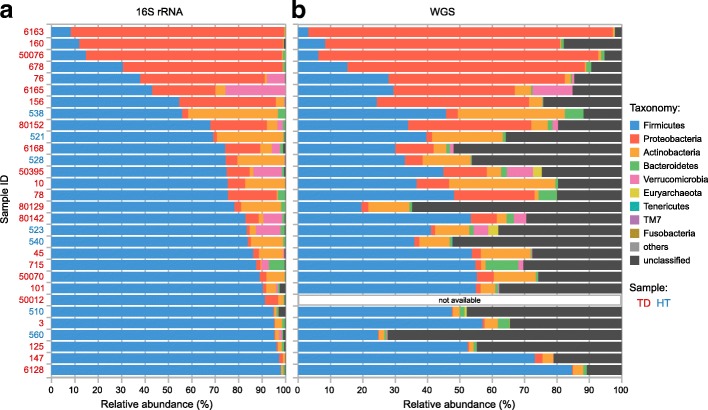
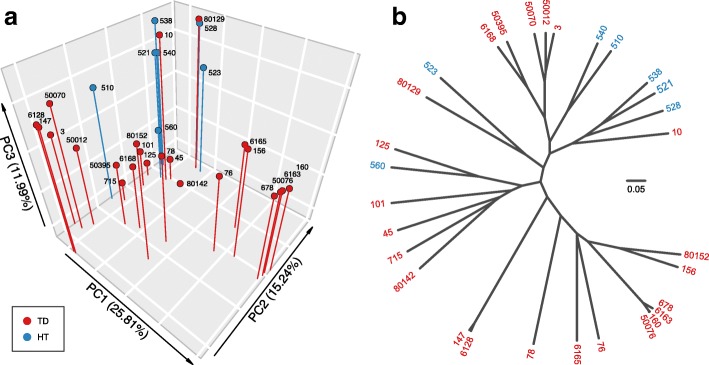
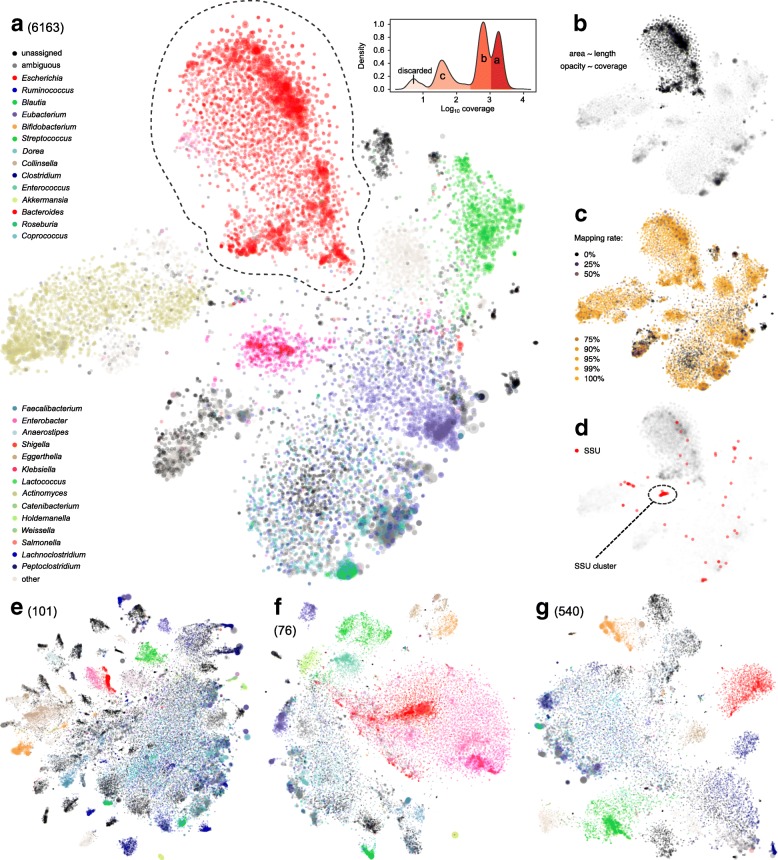
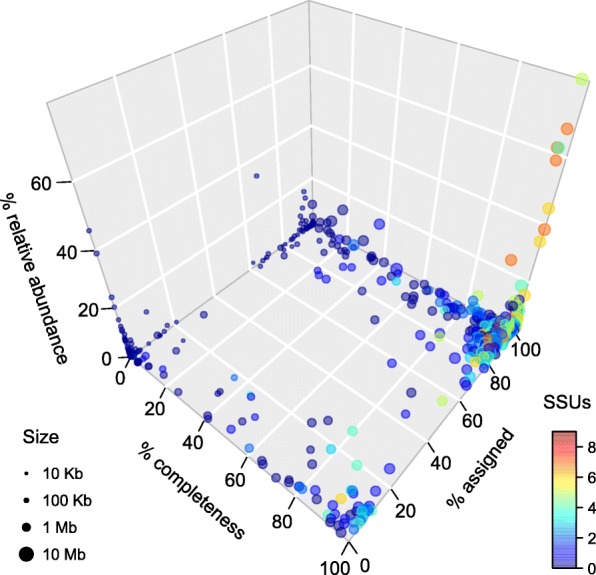
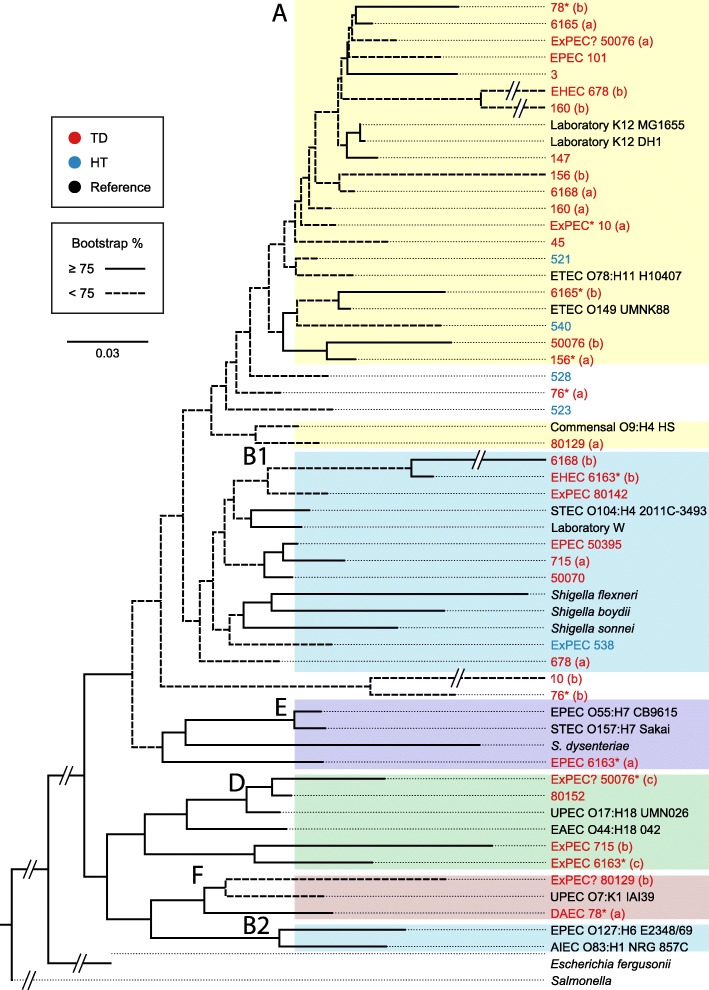
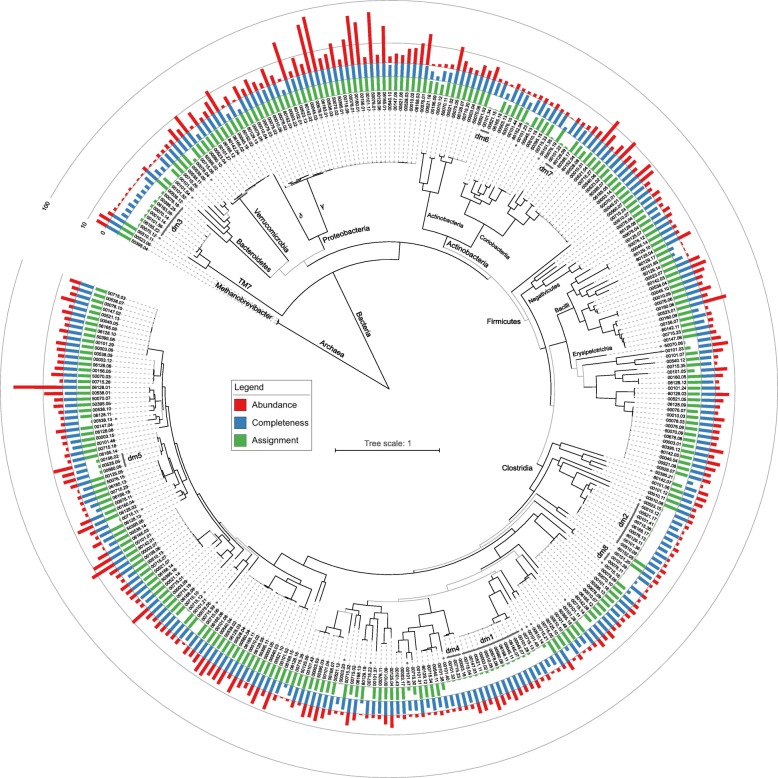
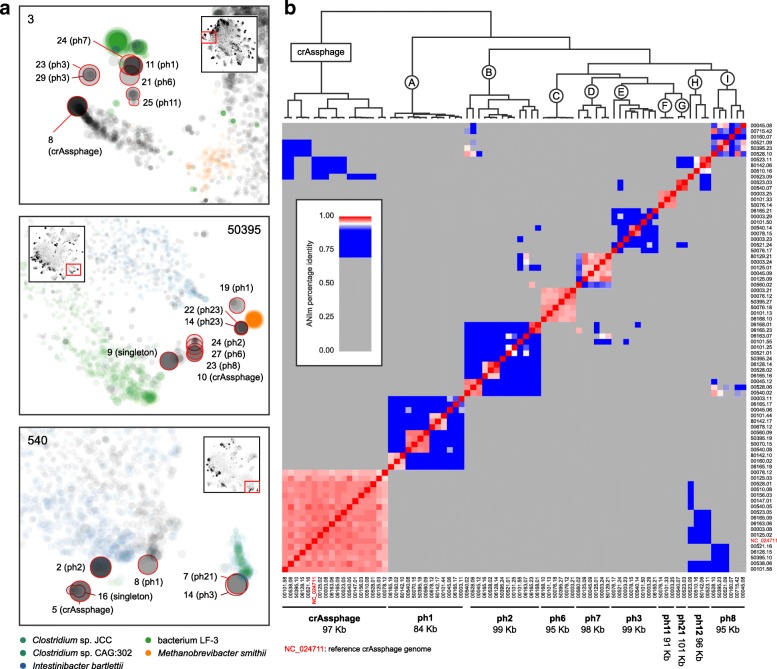
Results: We performed a comprehensive amplicon and whole genome shotgun (WGS) metagenomic study of the fecal microbiomes from 23 TD patients and seven healthy travelers, all of which were negative for the known etiologic agents of TD based on standard microbiological and immunological assays. Abnormal and diverse taxonomic profiles in TD samples were revealed. WGS reads were assembled and the resulting contigs were visualized using multiple query types. A semi-manual workflow was applied to isolate independent genomes from metagenomic pools. A total of 565 genome bins were extracted, 320 of which were complete enough to be characterized as cellular genomes; 160 were viral genomes. We made predictions of the etiology of disease for many of the individual subjects based on the properties and features of the recovered genomes. Multiple patients with low-diversity metagenomes were predominated by one to several E. coli strains. Functional annotation allowed prediction of pathogenic type in many cases. Five patients were co-infected with E. coli and other members of Enterobacteriaceae, including Enterobacter, Klebsiella, and Citrobacter; these may represent blooms of organisms that appear following secretory diarrhea. New "dark matter" microbes were observed in multiple samples. In one, we identified a novel TM7 genome that phylogenetically clustered with a sludge isolate; it carries genes encoding potential virulence factors. In multiple samples, we observed high proportions of putative novel viral genomes, some of which form clusters with the ubiquitous gut virus, crAssphage. The total relative abundance of viruses was significantly higher in healthy travelers versus TD patients.
Conclusion: Our study highlights the strength of assembly-based metagenomics, especially the manually curated, visualization-assisted binning of contigs, in resolving unusual and under-characterized pathogenic profiles of human-associated microbiomes. Results show that TD may be polymicrobial, with multiple novel cellular and viral strains as potential players in the diarrheal disease.
Keywords: Dark matter; Escherichia coli; Strain-level; TM7; Travelers’ diarrhea; Virulence factor; crAssphage.
Conflict of interest statement
Ethics approval and consent to participate
This project was granted an exemption from required ethics approval by the J. Craig Venter Institute Institutional Review Board.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
