

Quantifying the contribution of recessive coding variation to developmental disorders
- PMID: 30409806
- PMCID: PMC6726470
- DOI: 10.1126/science.aar6731
Quantifying the contribution of recessive coding variation to developmental disorders
Abstract
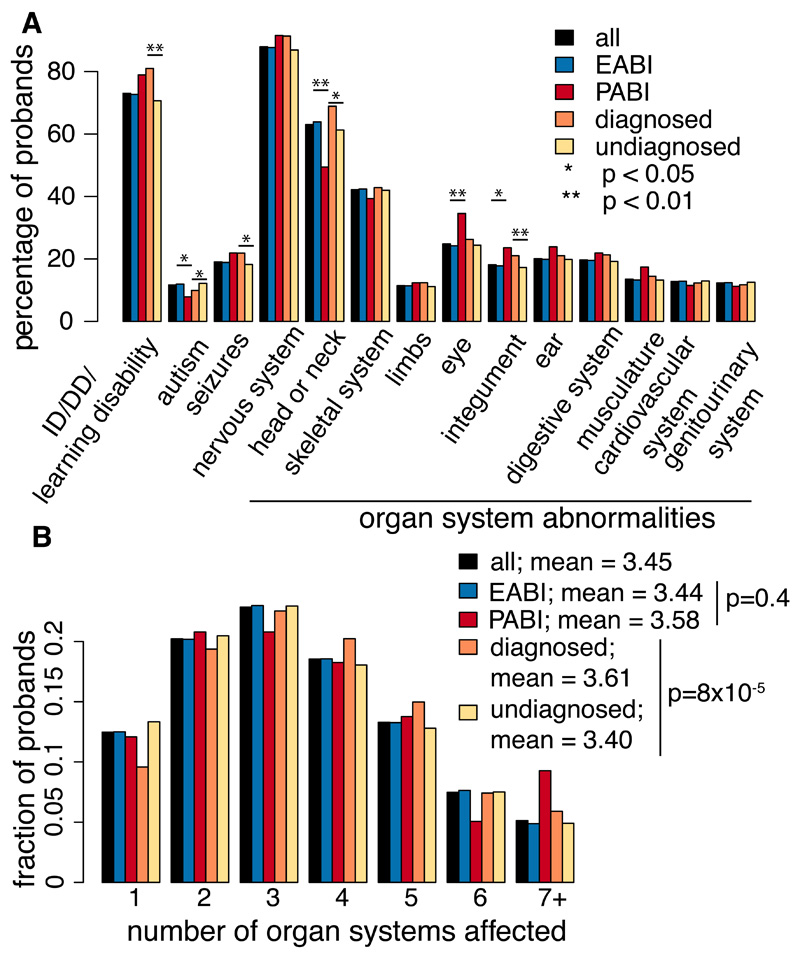
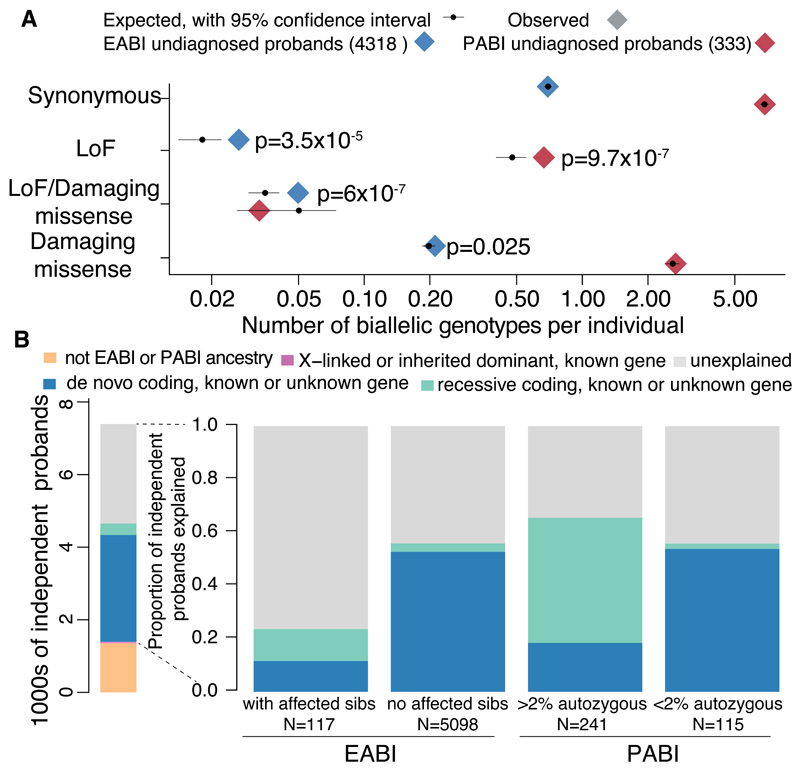
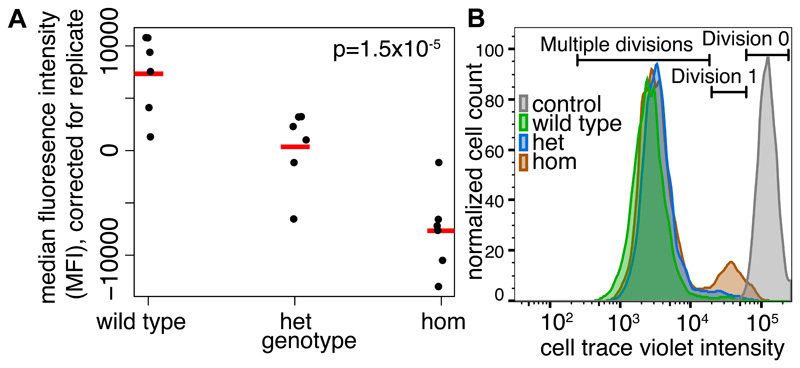
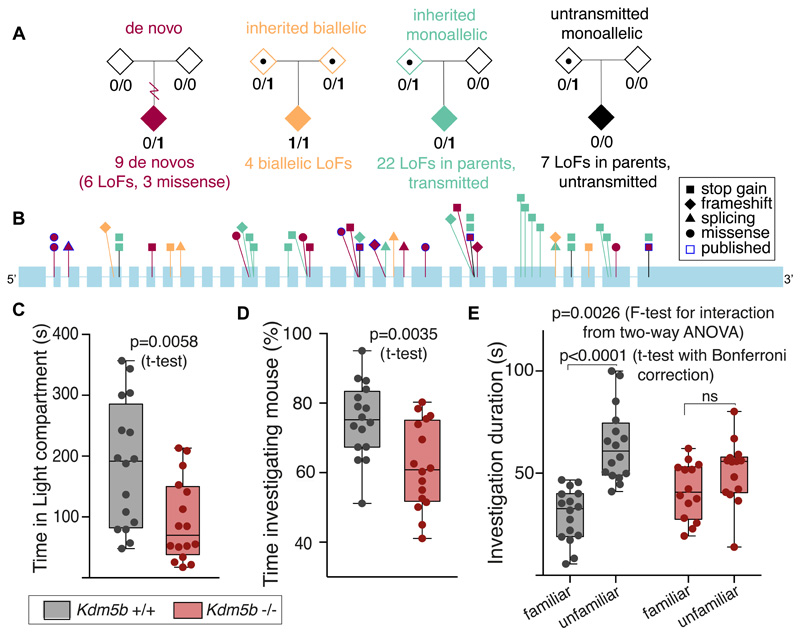
We estimated the genome-wide contribution of recessive coding variation in 6040 families from the Deciphering Developmental Disorders study. The proportion of cases attributable to recessive coding variants was 3.6% in patients of European ancestry, compared with 50% explained by de novo coding mutations. It was higher (31%) in patients with Pakistani ancestry, owing to elevated autozygosity. Half of this recessive burden is attributable to known genes. We identified two genes not previously associated with recessive developmental disorders, KDM5B and EIF3F, and functionally validated them with mouse and cellular models. Our results suggest that recessive coding variants account for a small fraction of currently undiagnosed nonconsanguineous individuals, and that the role of noncoding variants, incomplete penetrance, and polygenic mechanisms need further exploration.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
-
- Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–187. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
