

A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence
- PMID: 30409989
- PMCID: PMC6224596
- DOI: 10.1038/s41598-018-34812-x
A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence
Abstract
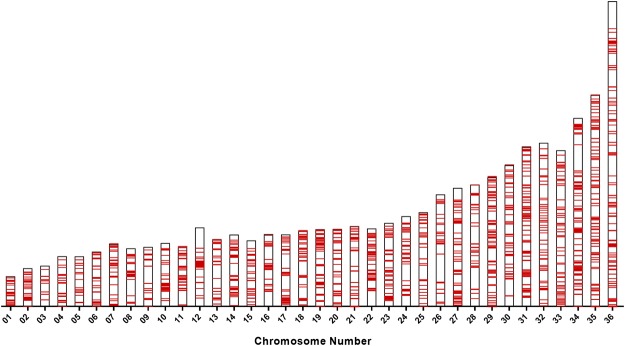
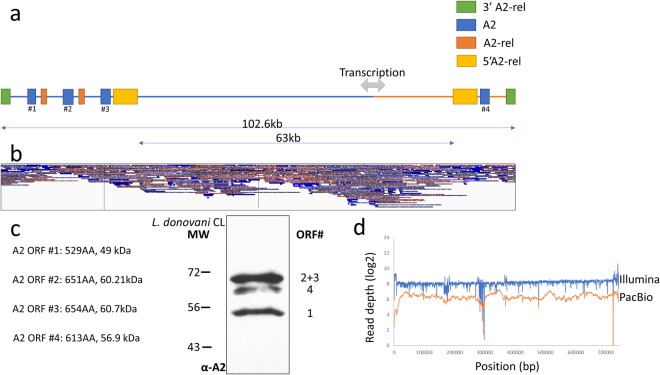
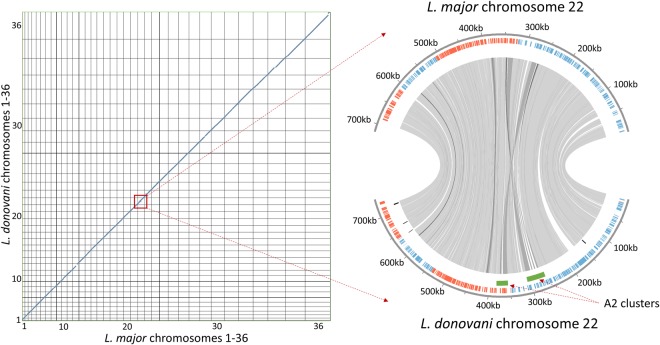
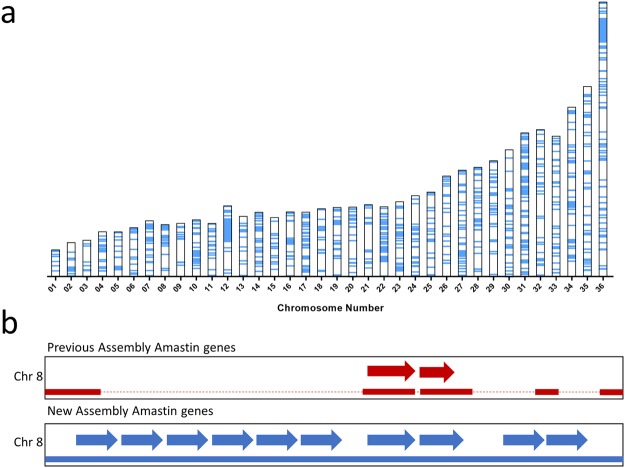
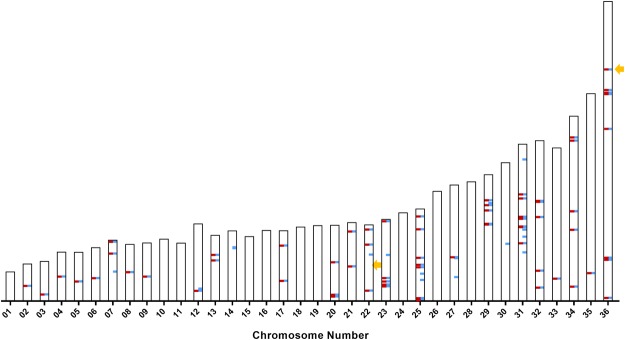
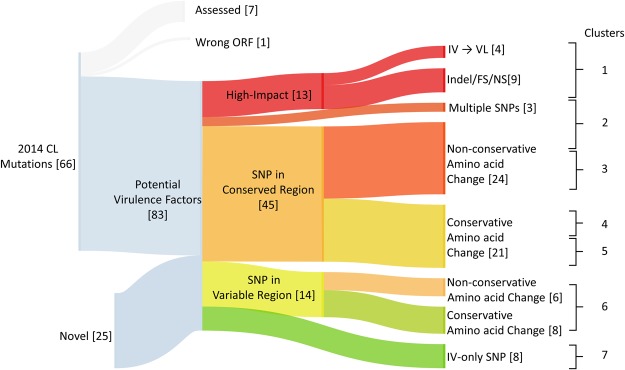
Leishmania donovani is responsible for visceral leishmaniasis, a neglected and lethal parasitic disease with limited treatment options and no vaccine. The study of L. donovani has been hindered by the lack of a high-quality reference genome and this can impact experimental outcomes including the identification of virulence genes, drug targets and vaccine development. We therefore generated a complete genome assembly by deep sequencing using a combination of second generation (Illumina) and third generation (PacBio) sequencing technologies. Compared to the current L. donovani assembly, the genome assembly reported within resulted in the closure over 2,000 gaps, the extension of several chromosomes up to telomeric repeats and the re-annotation of close to 15% of protein coding genes and the annotation of hundreds of non-coding RNA genes. It was possible to correctly assemble the highly repetitive A2 and Amastin virulence gene clusters. A comparative sequence analysis using the improved reference genome confirmed 70 published and identified 15 novel genomic differences between closely related visceral and atypical cutaneous disease-causing L. donovani strains providing a more complete map of genes associated with virulence and visceral organ tropism. Bioinformatic tools including protein variation effect analyzer and basic local alignment search tool were used to prioritize a list of potential virulence genes based on mutation severity, gene conservation and function. This complete genome assembly and novel information on virulence factors will support the identification of new drug targets and the development of a vaccine for L. donovani.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
