

GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures
- PMID: 30410121
- PMCID: PMC6681916
- DOI: 10.1038/nrd.2018.180
GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures
Abstract
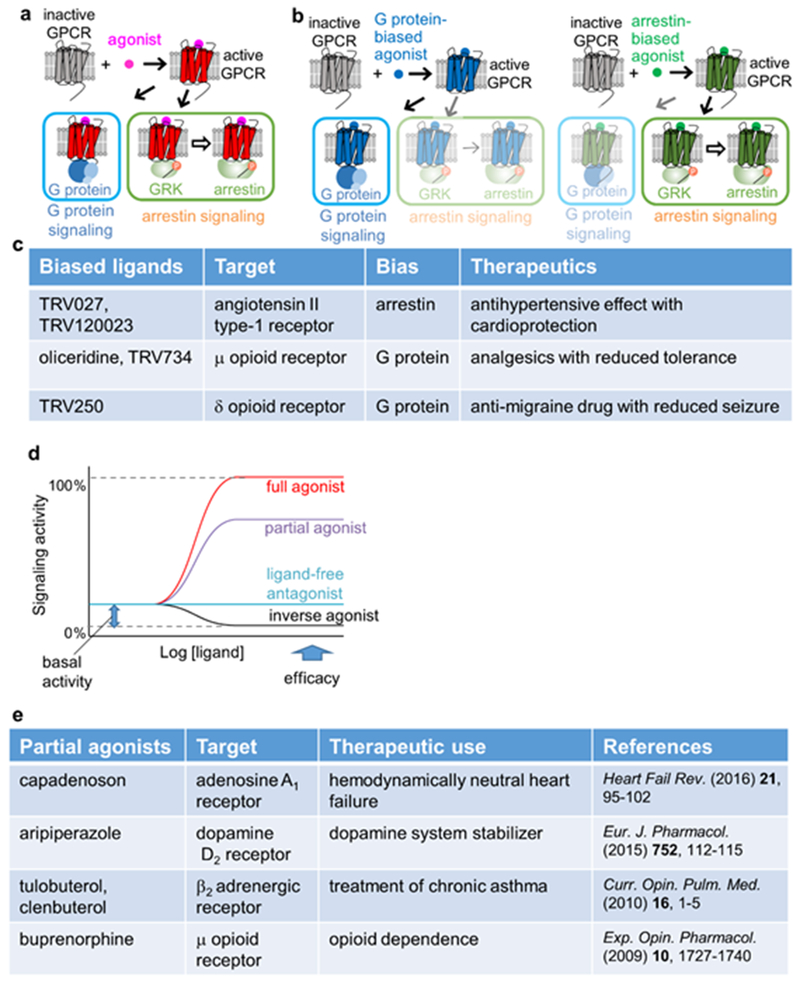
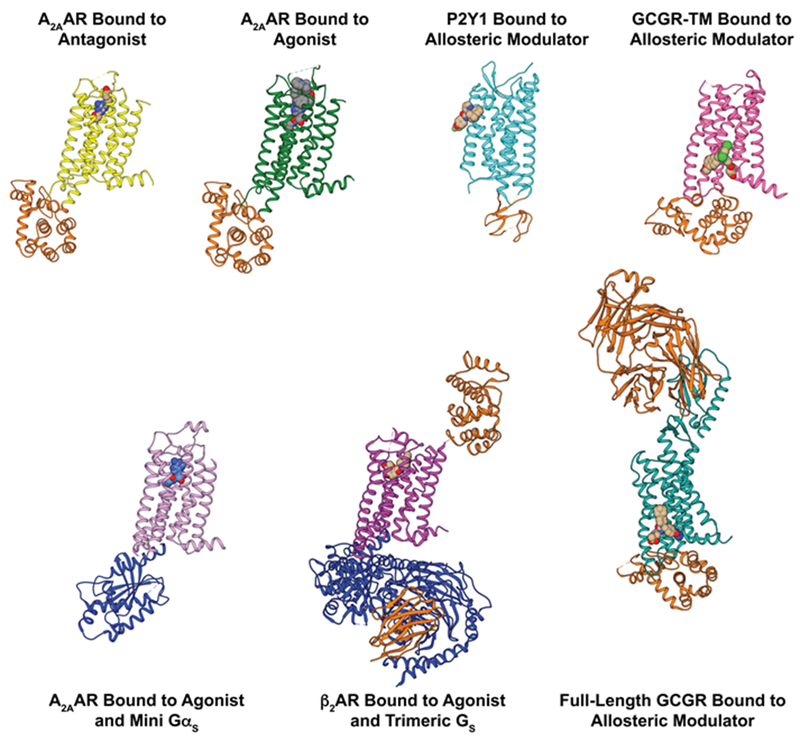
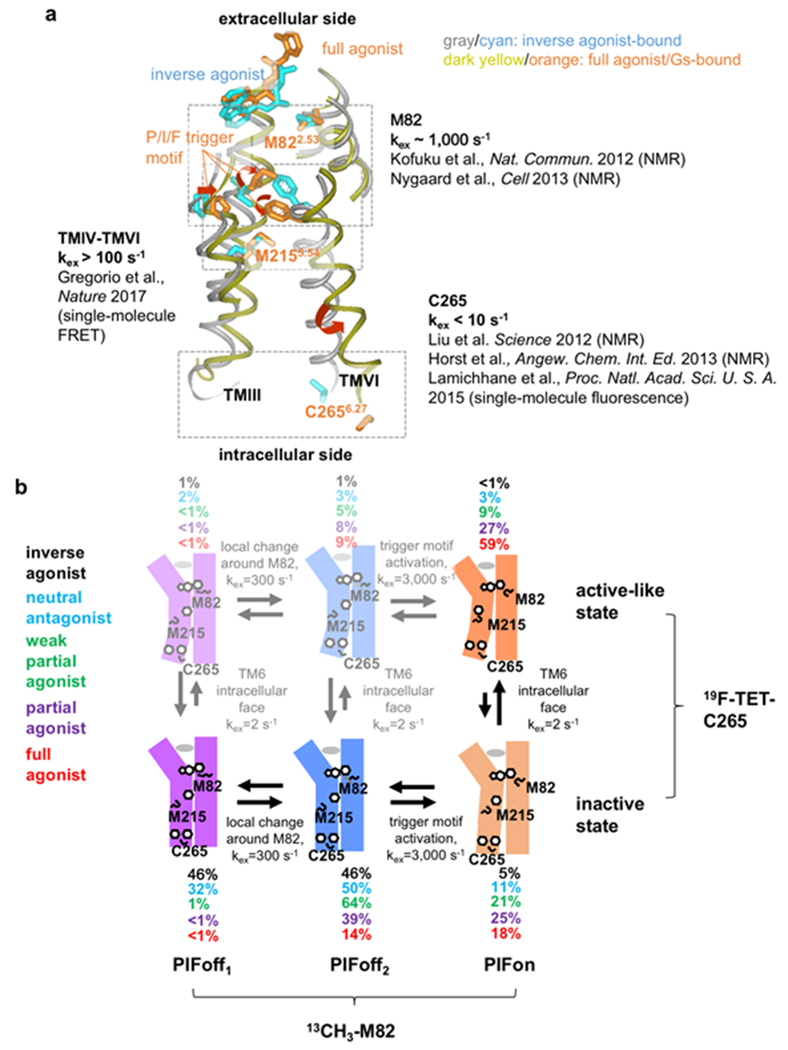
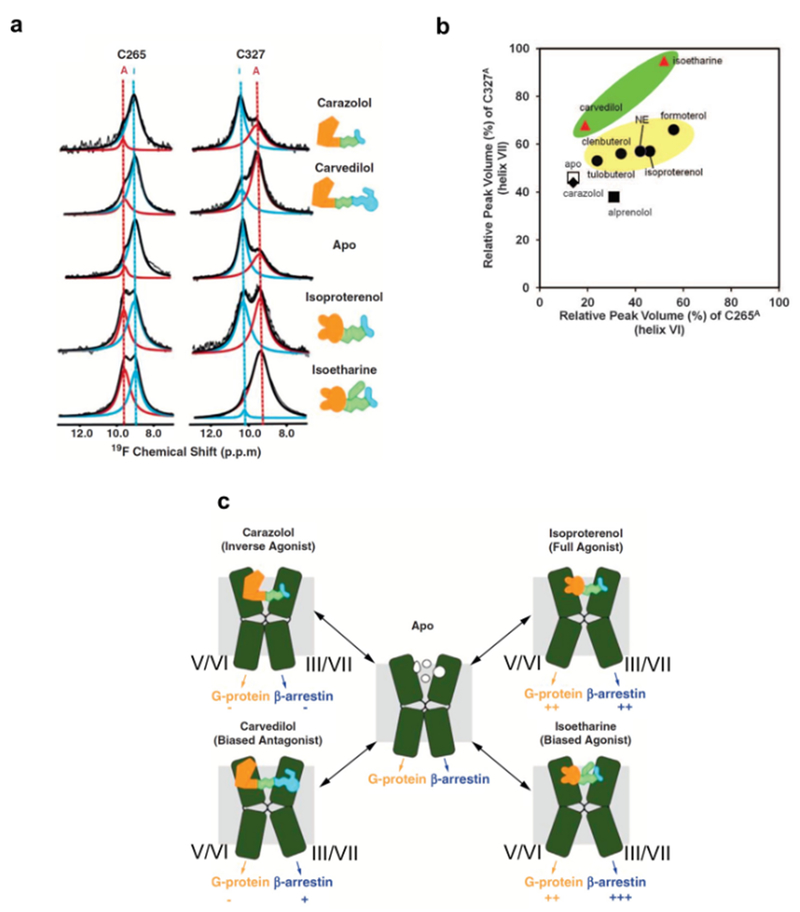
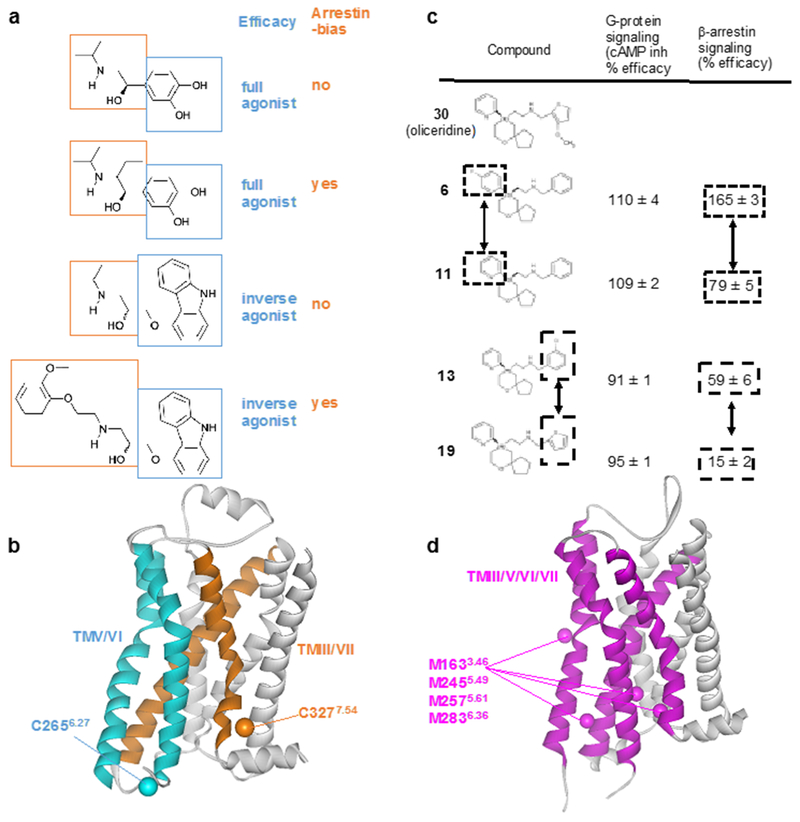
The 826 G protein-coupled receptors (GPCRs) in the human proteome regulate key physiological processes and thus have long been attractive drug targets. With the crystal structures of more than 50 different human GPCRs determined over the past decade, an initial platform for structure-based rational design has been established for drugs that target GPCRs, which is currently being augmented with cryo-electron microscopy (cryo-EM) structures of higher-order GPCR complexes. Nuclear magnetic resonance (NMR) spectroscopy in solution is one of the key approaches for expanding this platform with dynamic features, which can be accessed at physiological temperature and with minimal modification of the wild-type GPCR covalent structures. Here, we review strategies for the use of advanced biochemistry and NMR techniques with GPCRs, survey projects in which crystal or cryo-EM structures have been complemented with NMR investigations and discuss the impact of this integrative approach on GPCR biology and drug discovery.
Figures
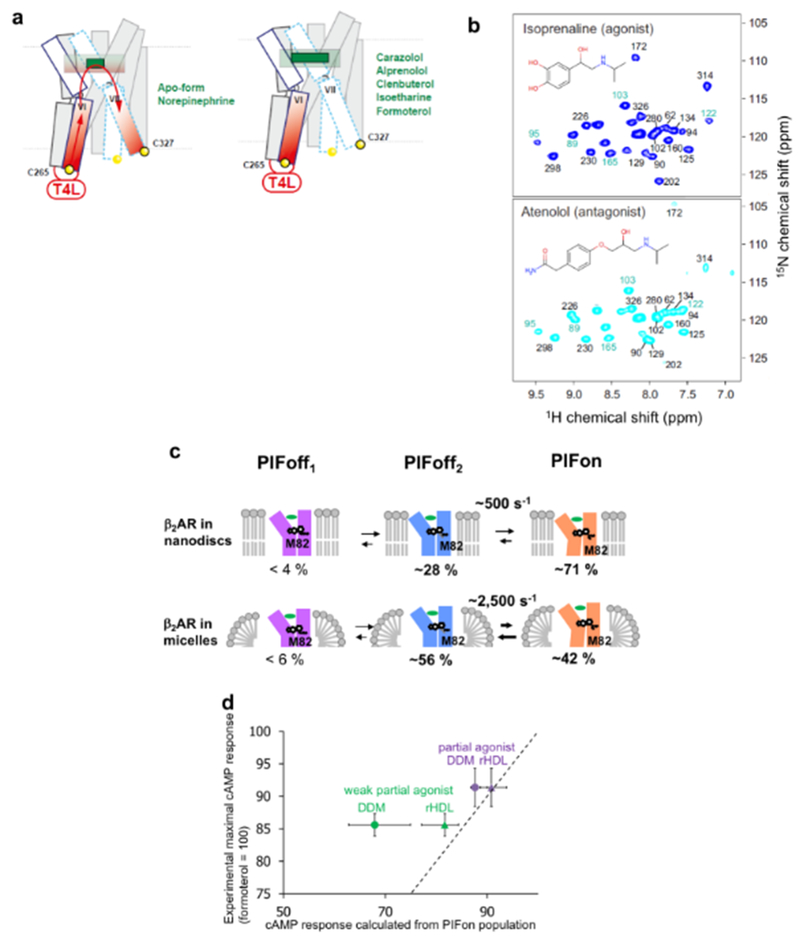
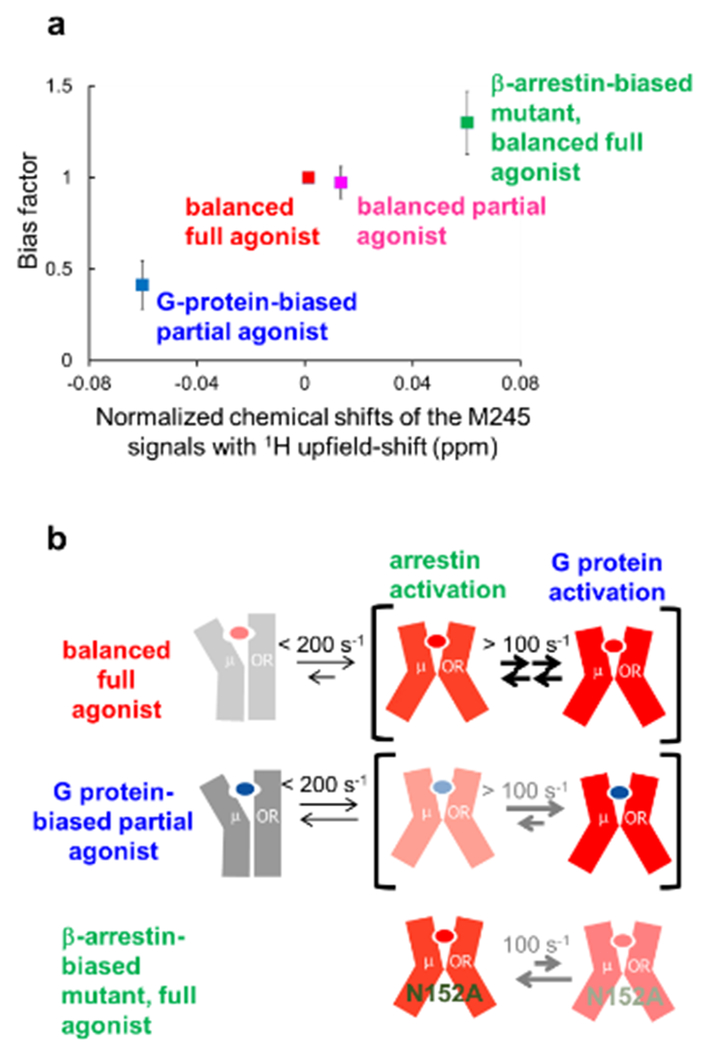
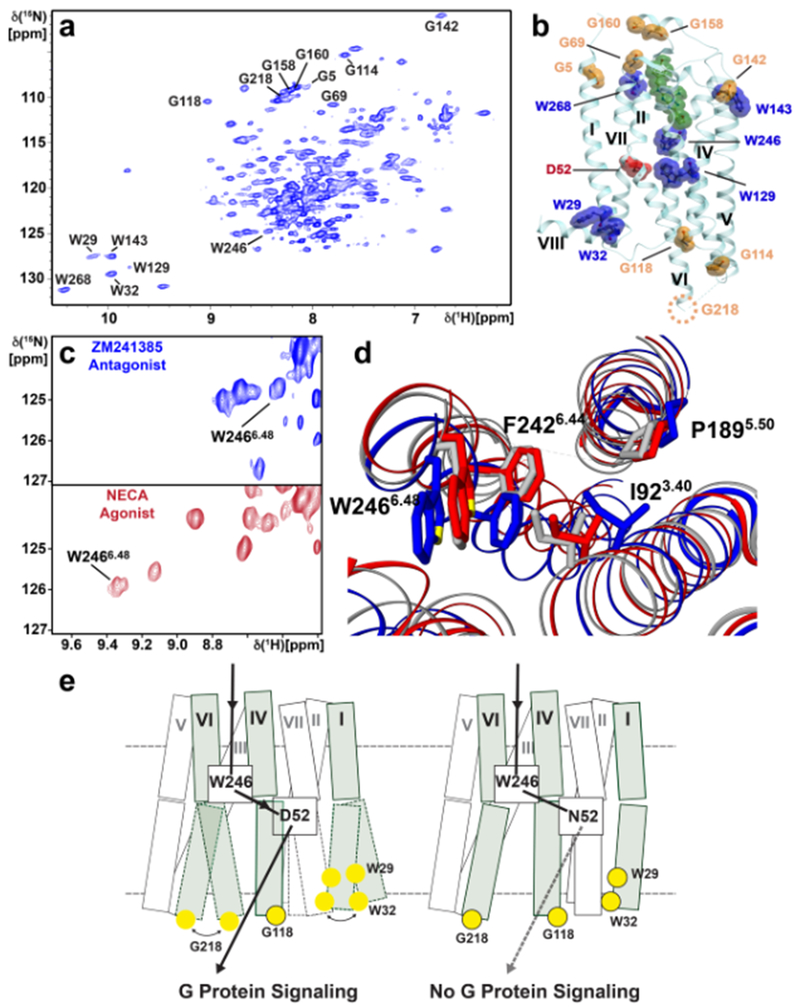
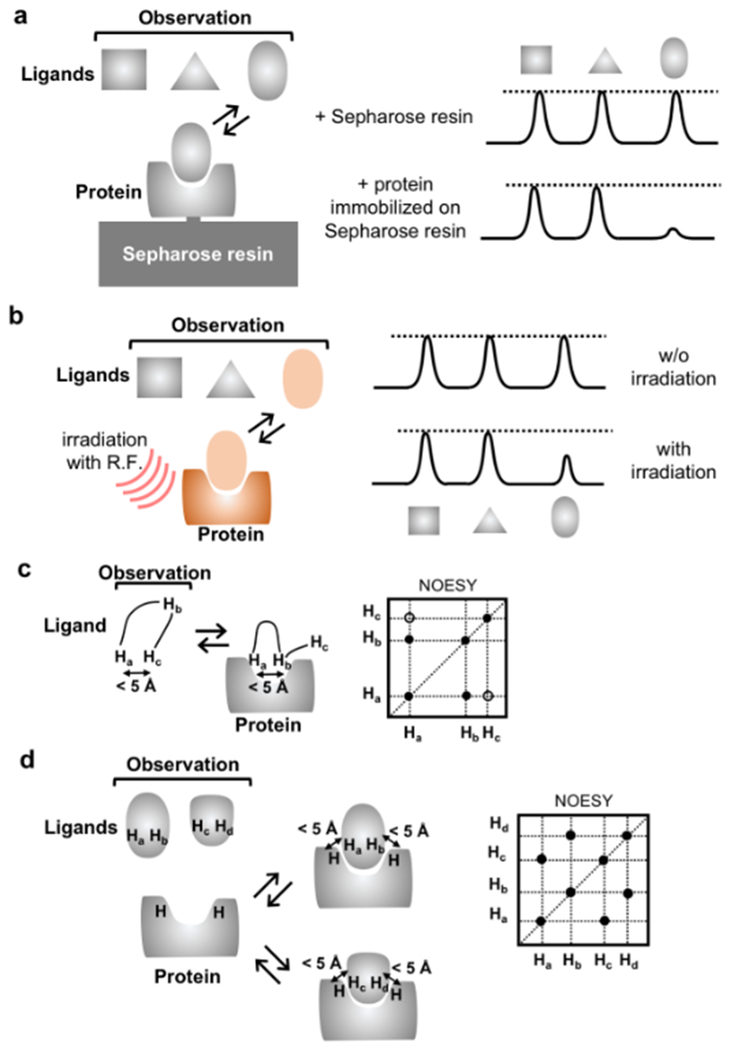
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
