

Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum
- PMID: 30415838
- PMCID: PMC6291210
- DOI: 10.1016/j.cell.2018.10.023
Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum
Abstract
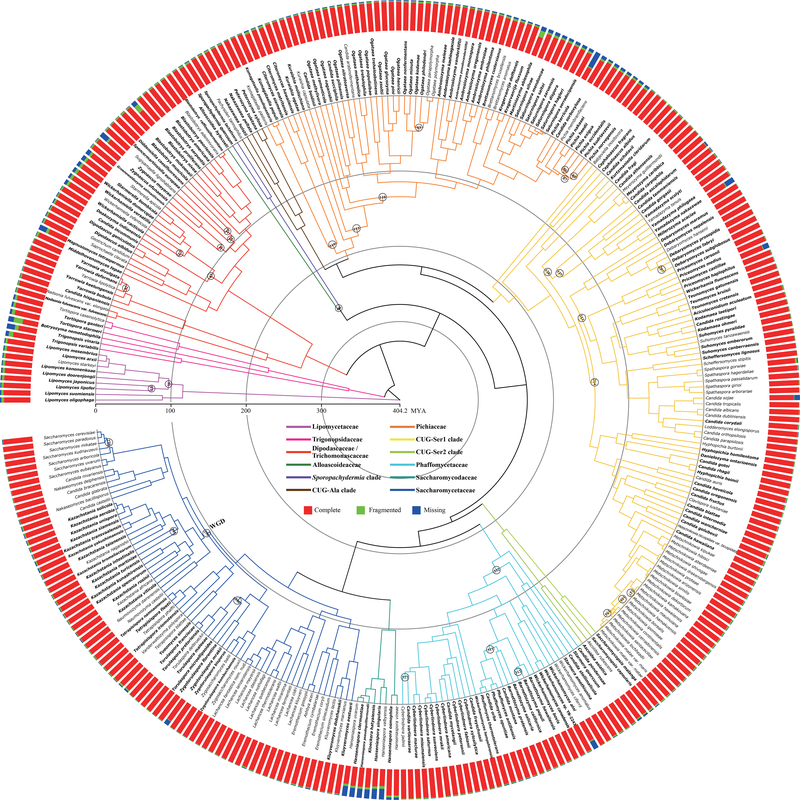
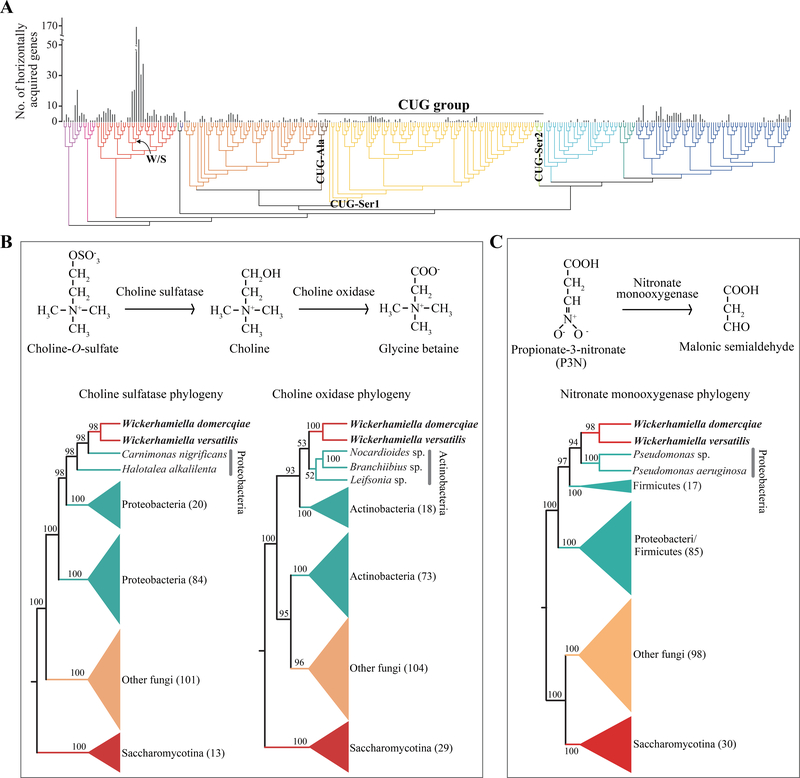
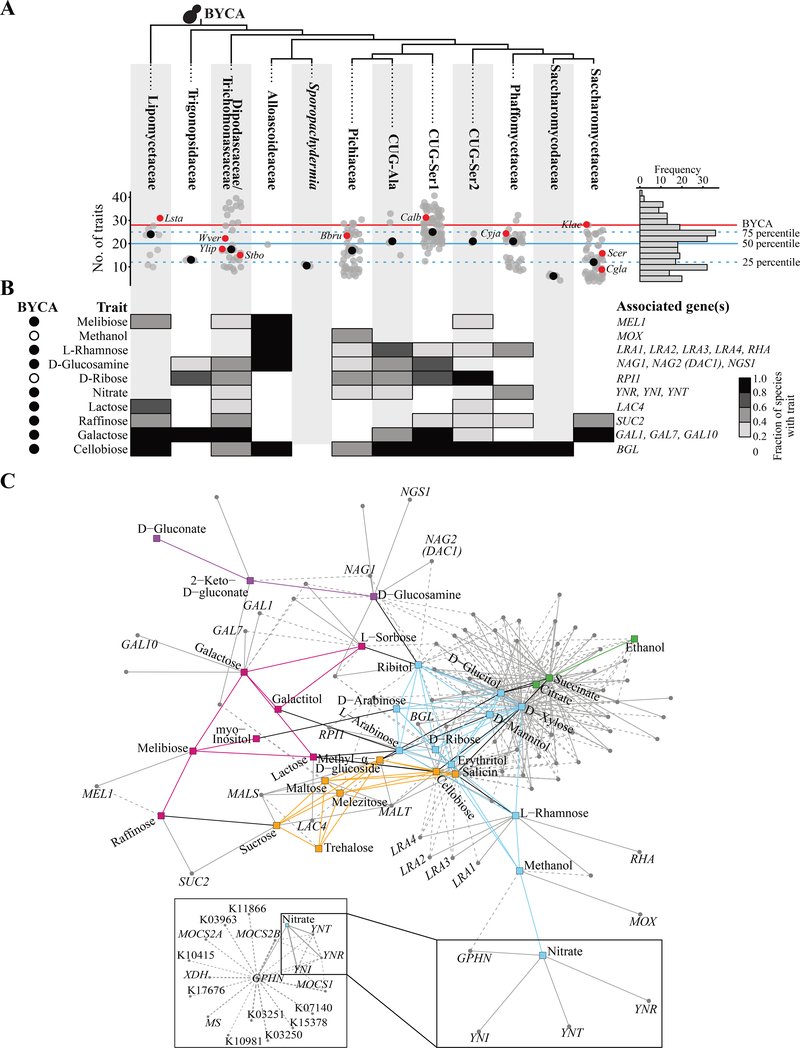
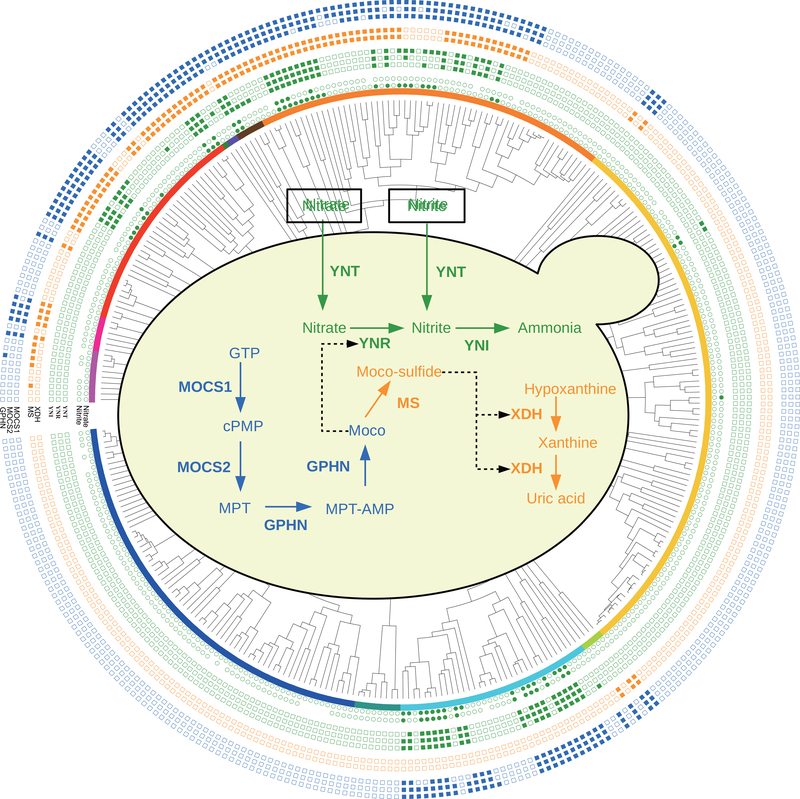
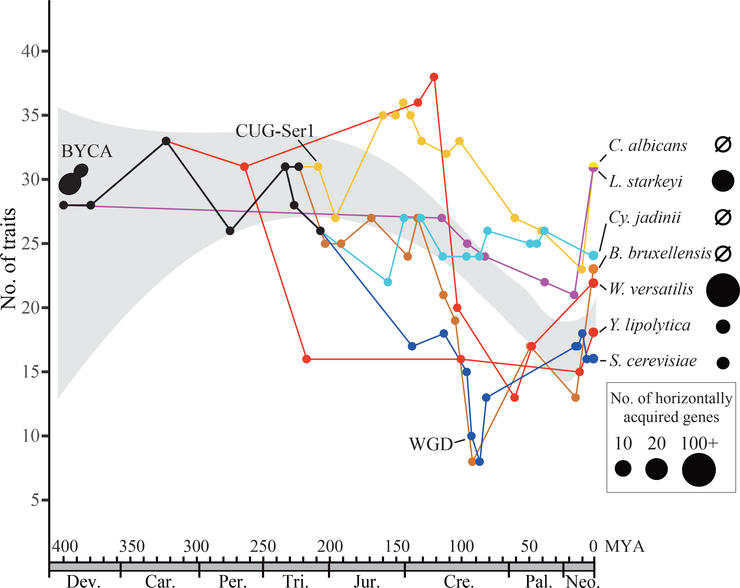
Budding yeasts (subphylum Saccharomycotina) are found in every biome and are as genetically diverse as plants or animals. To understand budding yeast evolution, we analyzed the genomes of 332 yeast species, including 220 newly sequenced ones, which represent nearly one-third of all known budding yeast diversity. Here, we establish a robust genus-level phylogeny comprising 12 major clades, infer the timescale of diversification from the Devonian period to the present, quantify horizontal gene transfer (HGT), and reconstruct the evolution of 45 metabolic traits and the metabolic toolkit of the budding yeast common ancestor (BYCA). We infer that BYCA was metabolically complex and chronicle the tempo and mode of genomic and phenotypic evolution across the subphylum, which is characterized by very low HGT levels and widespread losses of traits and the genes that control them. More generally, our results argue that reductive evolution is a major mode of evolutionary diversification.
Keywords: Ascomycota; Saccharomycotina; and reductive evolution; genomics; high-throughput sequencing; horizontal gene transfer; metabolic traits; molecular dating; phylogenetics; phylogenomics.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures

References
-
- Albalat R, and Canestro C (2016). Evolution by gene loss. Nat. Rev. Genet 17, 379–391. - PubMed
-
- Alexa A, and Rahnenfuhrer J (2016). Gene set enrichment analysis with topGO. Http://Www.Bioconductor.Org/.
-
- Bittner F, Oreb M, and Mendel RR (2001). ABA3 is a molybdenum cofactor sulfurase required for activation of aldehyde oxidase and xanthine dehydrogenase in Arabidopsis thaliana. J. Biol. Chem 276, 40381–40384. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
