

iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations
- PMID: 30417935
- PMCID: PMC6587874
- DOI: 10.1002/prot.25630
iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations
Abstract
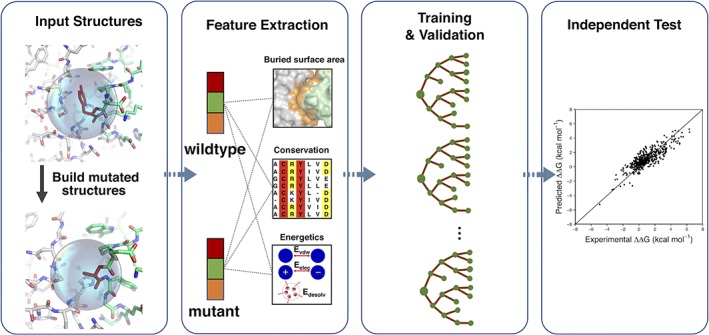
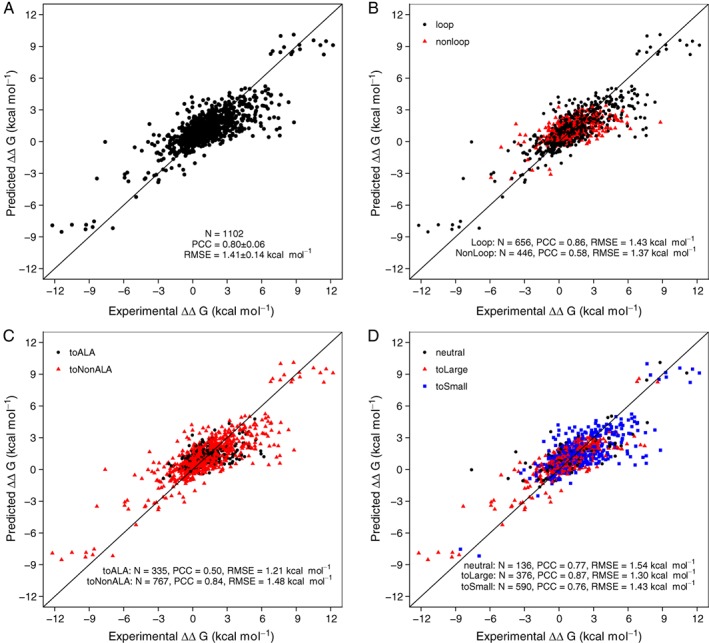
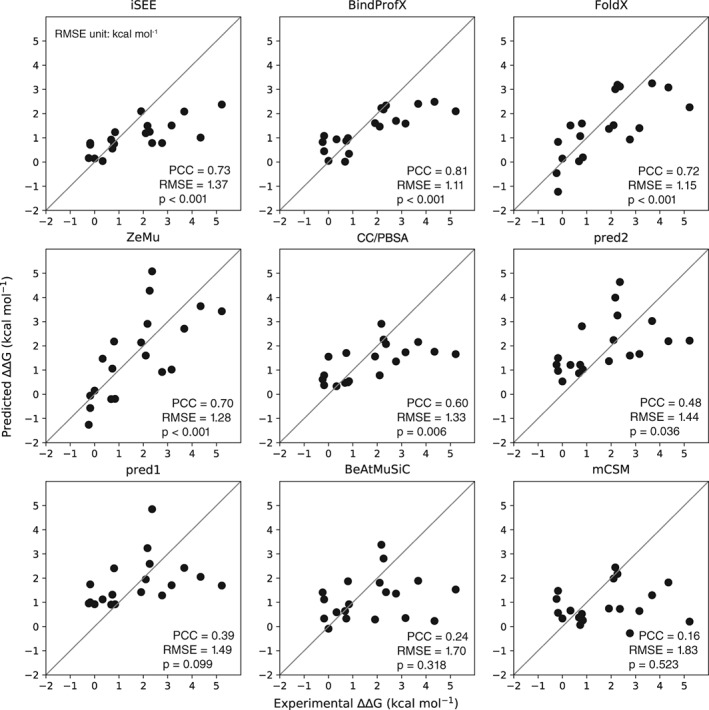
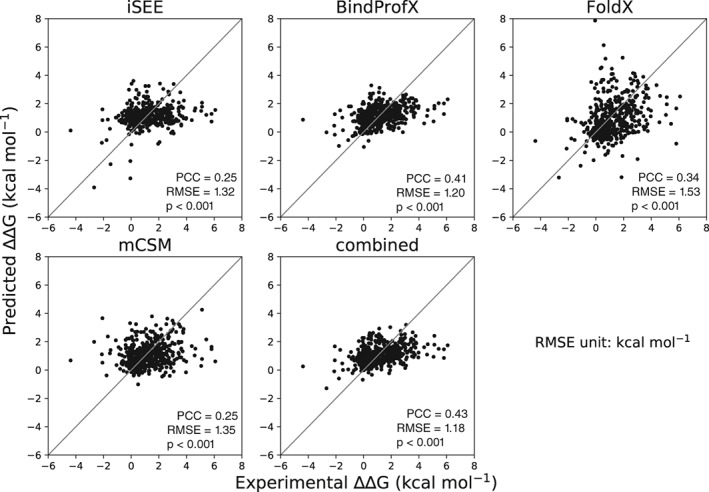
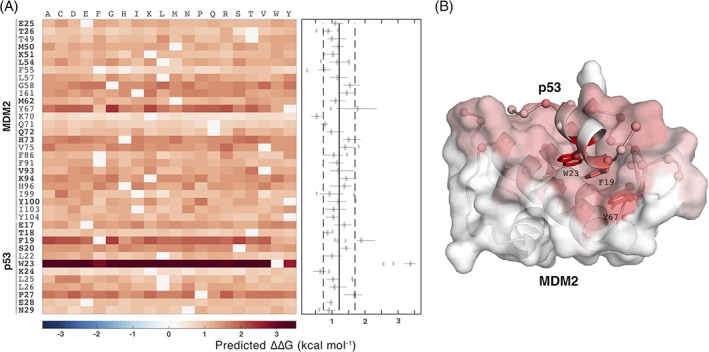
Quantitative evaluation of binding affinity changes upon mutations is crucial for protein engineering and drug design. Machine learning-based methods are gaining increasing momentum in this field. Due to the limited number of experimental data, using a small number of sensitive predictive features is vital to the generalization and robustness of such machine learning methods. Here we introduce a fast and reliable predictor of binding affinity changes upon single point mutation, based on a random forest approach. Our method, iSEE, uses a limited number of interface Structure, Evolution, and Energy-based features for the prediction. iSEE achieves, using only 31 features, a high prediction performance with a Pearson correlation coefficient (PCC) of 0.80 and a root mean square error of 1.41 kcal/mol on a diverse training dataset consisting of 1102 mutations in 57 protein-protein complexes. It competes with existing state-of-the-art methods on two blind test datasets. Predictions for a new dataset of 487 mutations in 56 protein complexes from the recently published SKEMPI 2.0 database reveals that none of the current methods perform well (PCC < 0.42), although their combination does improve the predictions. Feature analysis for iSEE underlines the significance of evolutionary conservations for quantitative prediction of mutation effects. As an application example, we perform a full mutation scanning of the interface residues in the MDM2-p53 complex.
Keywords: binding affinity; full mutation scanning; machine learning; protein-protein interactions; single point mutation.
© 2018 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals, Inc.
Figures
Similar articles
-
Analysis of the TP53 Deleterious Single Nucleotide Polymorphisms Impact on Estrogen Receptor Alpha-p53 Interaction: A Machine Learning Approach.Int J Mol Sci. 2019 Jun 18;20(12):2962. doi: 10.3390/ijms20122962. Int J Mol Sci. 2019. PMID: 31216622 Free PMC article.
-
Decomposing Structural Response Due to Sequence Changes in Protein Domains with Machine Learning.J Mol Biol. 2020 Jul 24;432(16):4435-4446. doi: 10.1016/j.jmb.2020.05.021. Epub 2020 May 30. J Mol Biol. 2020. PMID: 32485208
-
Assessing the performance of computational predictors for estimating protein stability changes upon missense mutations.Brief Bioinform. 2021 Nov 5;22(6):bbab184. doi: 10.1093/bib/bbab184. Brief Bioinform. 2021. PMID: 34058752
-
Machine Learning Approaches for Protein⁻Protein Interaction Hot Spot Prediction: Progress and Comparative Assessment.Molecules. 2018 Oct 4;23(10):2535. doi: 10.3390/molecules23102535. Molecules. 2018. PMID: 30287797 Free PMC article. Review.
-
Structural and sequential context of p53: A review of experimental and theoretical evidence.Prog Biophys Mol Biol. 2015 Mar;117(2-3):250-263. doi: 10.1016/j.pbiomolbio.2014.12.002. Epub 2014 Dec 27. Prog Biophys Mol Biol. 2015. PMID: 25550083 Review.
Cited by
-
Biomolecular Topology: Modelling and Analysis.Acta Math Sin Engl Ser. 2022;38(10):1901-1938. doi: 10.1007/s10114-022-2326-5. Epub 2022 Oct 15. Acta Math Sin Engl Ser. 2022. PMID: 36407804 Free PMC article.
-
Inferring the Effects of Protein Variants on Protein-Protein Interactions with Interpretable Transformer Representations.Research (Wash D C). 2023 Sep 11;6:0219. doi: 10.34133/research.0219. eCollection 2023. Research (Wash D C). 2023. PMID: 37701056 Free PMC article.
-
Mutation effect estimation on protein-protein interactions using deep contextualized representation learning.NAR Genom Bioinform. 2020 Jun;2(2):lqaa015. doi: 10.1093/nargab/lqaa015. Epub 2020 Mar 5. NAR Genom Bioinform. 2020. PMID: 32166223 Free PMC article.
-
DDMut-PPI: predicting effects of mutations on protein-protein interactions using graph-based deep learning.Nucleic Acids Res. 2024 Jul 5;52(W1):W207-W214. doi: 10.1093/nar/gkae412. Nucleic Acids Res. 2024. PMID: 38783112 Free PMC article.
-
Graph masked self-distillation learning for prediction of mutation impact on protein-protein interactions.Commun Biol. 2024 Oct 26;7(1):1400. doi: 10.1038/s42003-024-07066-9. Commun Biol. 2024. PMID: 39462102 Free PMC article.
References
-
- Steinbrecher T, Abel R, Clark A, Friesner R. Free energy perturbation calculations of the thermodynamics of protein side‐chain mutations. J Mol Biol. 2017;429(7):923‐929. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
