

Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel-associated retinopathy
- PMID: 30418171
- PMCID: PMC6264655
- DOI: 10.1172/JCI96098
Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel-associated retinopathy
Abstract
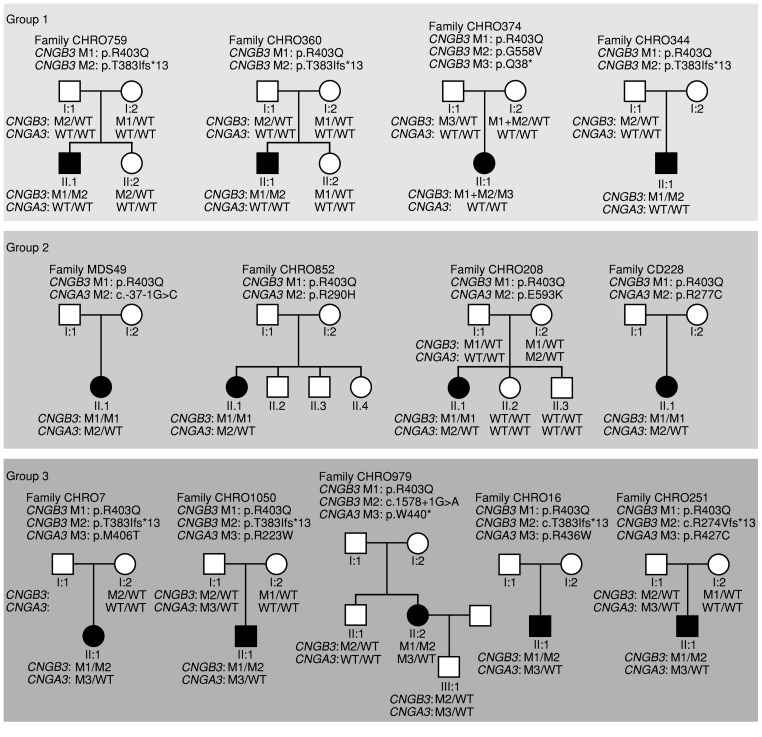
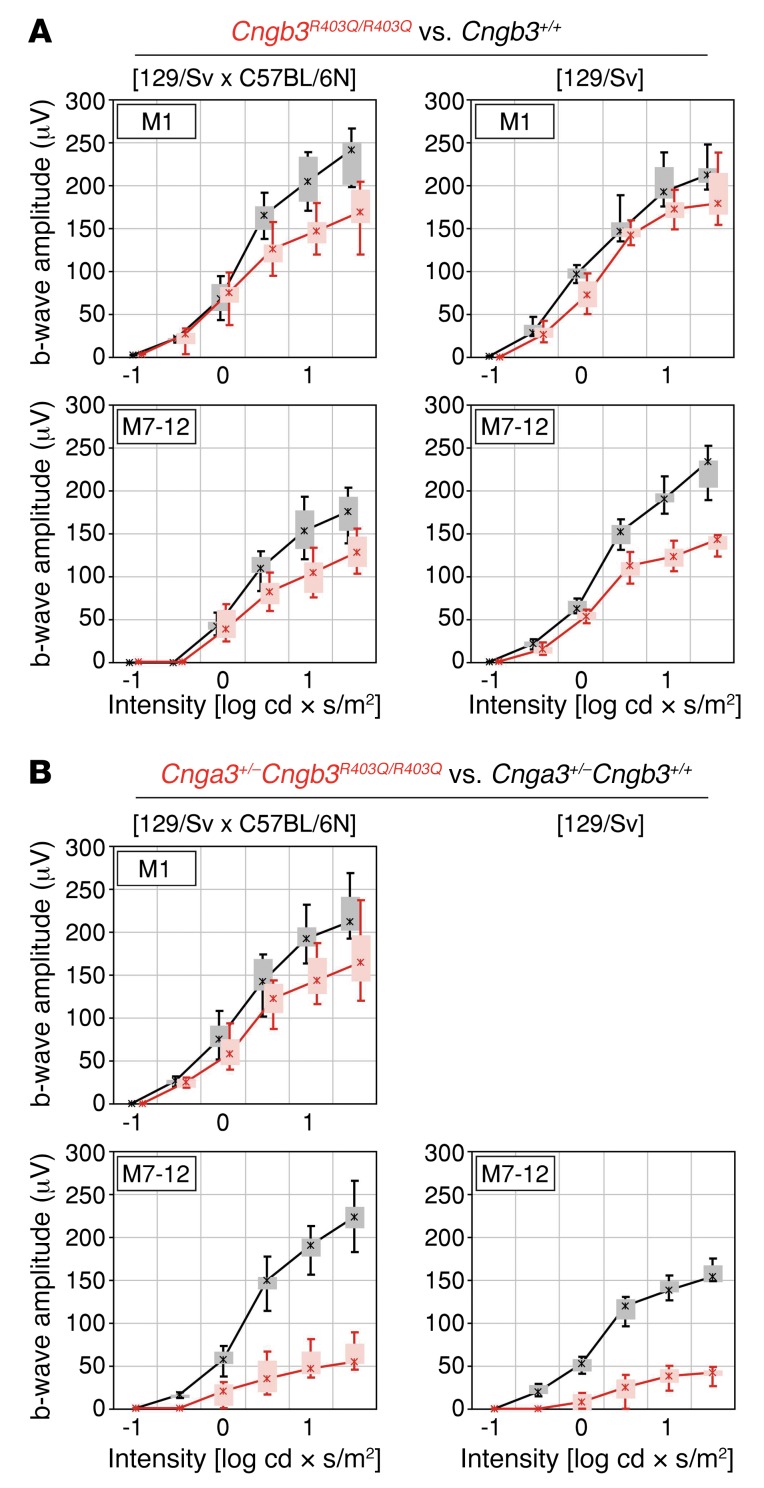
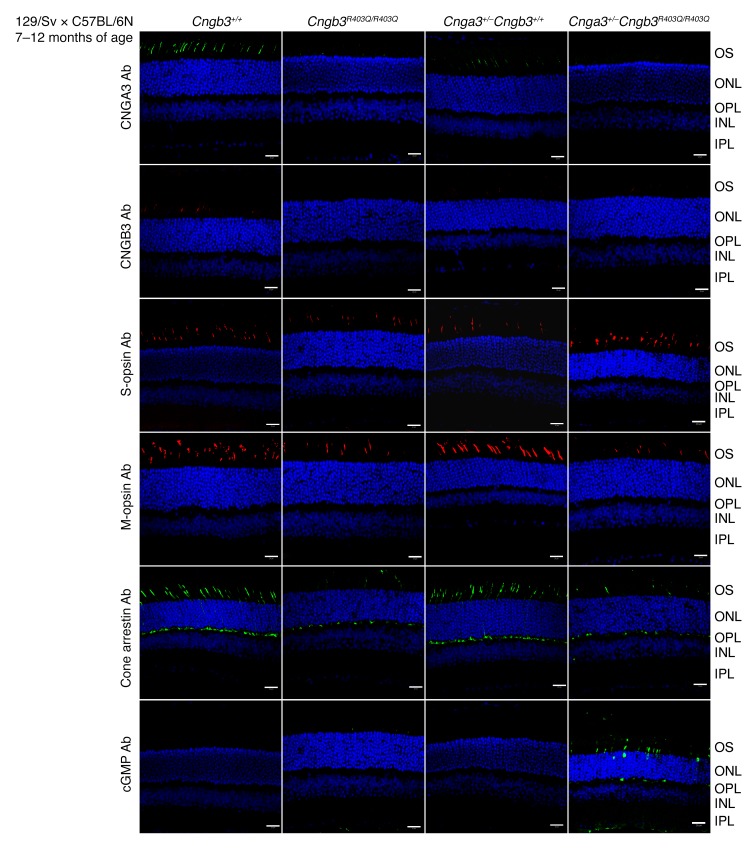
Mutations in CNGA3 and CNGB3, the genes encoding the subunits of the tetrameric cone photoreceptor cyclic nucleotide-gated ion channel, cause achromatopsia, a congenital retinal disorder characterized by loss of cone function. However, a small number of patients carrying the CNGB3/c.1208G>A;p.R403Q mutation present with a variable retinal phenotype ranging from complete and incomplete achromatopsia to moderate cone dysfunction or progressive cone dystrophy. By exploring a large patient cohort and published cases, we identified 16 unrelated individuals who were homozygous or (compound-)heterozygous for the CNGB3/c.1208G>A;p.R403Q mutation. In-depth genetic and clinical analysis revealed a co-occurrence of a mutant CNGA3 allele in a high proportion of these patients (10 of 16), likely contributing to the disease phenotype. To verify these findings, we generated a Cngb3R403Q/R403Q mouse model, which was crossbred with Cnga3-deficient (Cnga3-/-) mice to obtain triallelic Cnga3+/- Cngb3R403Q/R403Q mutants. As in human subjects, there was a striking genotype-phenotype correlation, since the presence of 1 Cnga3-null allele exacerbated the cone dystrophy phenotype in Cngb3R403Q/R403Q mice. These findings strongly suggest a digenic and triallelic inheritance pattern in a subset of patients with achromatopsia/severe cone dystrophy linked to the CNGB3/p.R403Q mutation, with important implications for diagnosis, prognosis, and genetic counseling.
Keywords: Genetics; Molecular genetics; Ophthalmology; Retinopathy.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
