

Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration
- PMID: 30420557
- PMCID: PMC6276871
- DOI: 10.15252/embj.2018100540
Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration
Abstract
A set of glutamylases and deglutamylases controls levels of tubulin polyglutamylation, a prominent post-translational modification of neuronal microtubules. Defective tubulin polyglutamylation was first linked to neurodegeneration in the Purkinje cell degeneration (pcd) mouse, which lacks deglutamylase CCP1, displays massive cerebellar atrophy, and accumulates abnormally glutamylated tubulin in degenerating neurons. We found biallelic rare and damaging variants in the gene encoding CCP1 in 13 individuals with infantile-onset neurodegeneration and confirmed the absence of functional CCP1 along with dysregulated tubulin polyglutamylation. The human disease mainly affected the cerebellum, spinal motor neurons, and peripheral nerves. We also demonstrate previously unrecognized peripheral nerve and spinal motor neuron degeneration in pcd mice, which thus recapitulated key features of the human disease. Our findings link human neurodegeneration to tubulin polyglutamylation, entailing this post-translational modification as a potential target for drug development for neurodegenerative disorders.
Keywords: cerebellum; cytosolic carboxypeptidase 1 (CCP1/AGTPBP1/NNA1); motor neuron; neurodegeneration; tubulin polyglutamylation.
© 2018 The Authors.
Figures
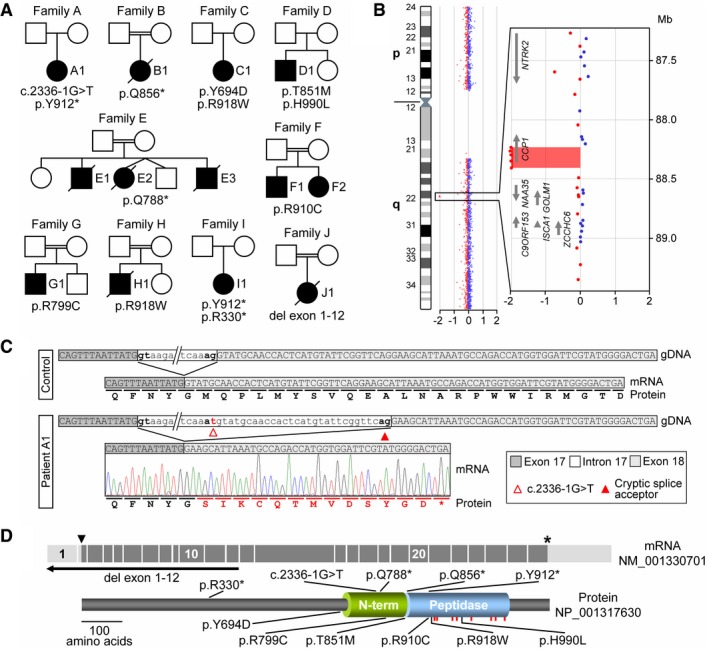
Pedigrees of ten families with affected individuals carrying biallelic variants identified by WES or array CGH.
Array CGH ratio profile of chromosome 9 from patient J1. Left: chromosome 9 ideogram with the log2ratios of probes plotted as a function of chromosomal position. Right: zoomed‐in region of interest containing the presumed homozygous deletion encompassing the N‐terminal exons of the CCP1 gene. Arrows point in the direction of transcription.
Characterization of the splice acceptor site variant in family A. At the mRNA level, the intron 17 variant c.2336‐1G >T resulted in activation of a cryptic splice site, removing 29 nt from the mRNA causing a frameshift (p.M780fs) of the CCP1 open reading frame.
Schematic representation of the mRNA (exons 1–26), the encoded CCP1 protein, and positions of disease‐associated variants. Arrowhead: ATG start codon, asterisk: stop codon. Peptidase: carboxypeptidase domain, N‐term: conserved N‐terminal domain, red bars: localizations of residues required for CCP1 substrate binding and catalytic activity.
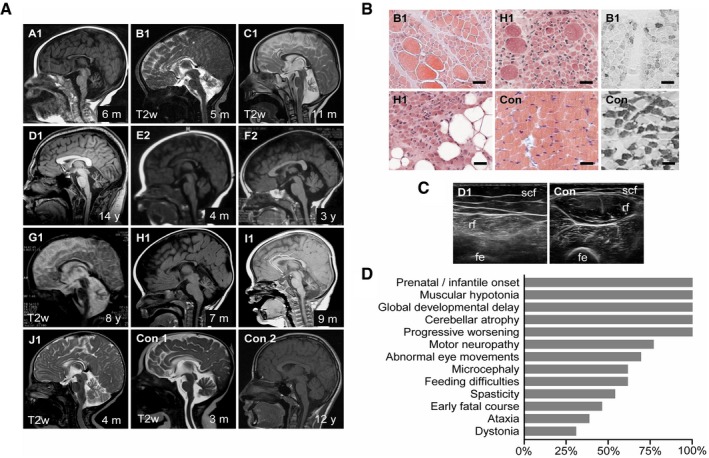
Cerebellar atrophy (severe: A1, B1, C1, D1, G1, H1, I1; moderate: E2, F2, J1) and corpus callosum dysplasia (A1, B1, E2, J1). Magnetic resonance scans, T1w except where indicated. Con1‐2: non‐disease controls. Age at examination is given in months (m) or years (y).
Chronic denervation with group fiber atrophy, interspersed hypertrophic fibers and fatty replacement (hematoxylin–eosin staining, left), and type 1 fiber predominance (fast myosin heavy‐chain staining, right). M. quadriceps biopsies at age 5 (B1) and 7 months (H1). Con: non‐disease control. Scale bar, 50 μm.
Chronic denervation indicated by atrophy and increased echogenicity with granular and streaky pattern. M. rectus femoris (rf) ultrasound at age 14 years (D1). Con: healthy individual. Scf: subcutaneous fat, fe: femur.
Relative frequencies of signs and symptoms of CCP1‐associated neurodegeneration based on 13 affected individuals. For details, see Appendix Table S4.
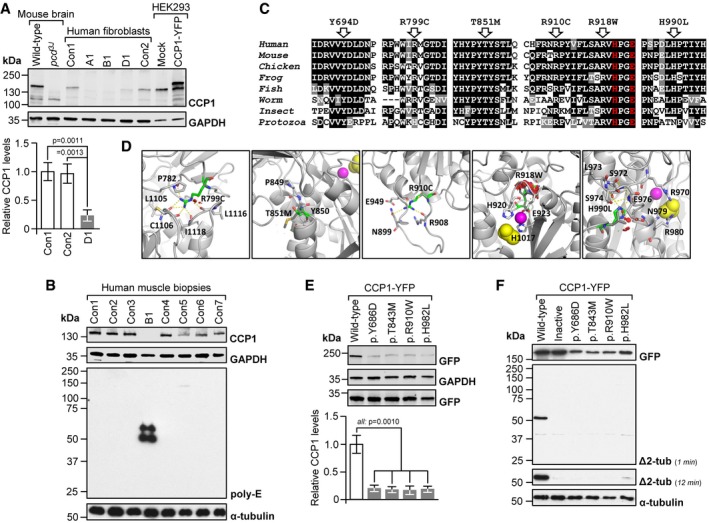
Absent (A1, B1) or low‐level CCP1 (D1) in patient‐derived skin fibroblasts. Con1‐2: healthy donors. No signal in pcd brain and a higher‐molecular‐weight band in CCP1‐YFP‐expressing cells demonstrated specificity of the CCP1 antibody. Graphs: mean ± SD, n = 3. P‐values: one‐way ANOVA, Tukey post‐test.
Excess tubulin polyglutamylation in CCP1‐deficient human skeletal muscle (M. quadriceps). B1: individual B1, Con1‐7: non‐disease controls. Poly‐E: polyglutamylated tubulin generated through the activity of polyglutamylases. Presence of CCP1 in human muscle was confirmed using the anti‐CCP1 antibody.
Alignments of partial sequences of CCP1 from multiple species. Arrows: positions of CCP1 missense mutations observed in patients. Residues printed in red are required for enzymatic activity. Human (H. sapiens): NP_001317630, mouse (M. musculus): NP_075817, chicken (G. gallus): NP_001292036, frog (X. laevis): XP_018099357, fish (D. rerio): NP_001019616, worm (C. elegans): NP_491674, insect (A. mellifera): XP_006571511, and protozoa (T. brucei): XP_011778470.
Possible effects of missense mutants on a predicted 3D structure of human CCP1 (based on 4b6z). Green stick models: mutated residues, sphere models: zinc ion (magenta) and acetate ion (substrate mimic, yellow), and dashed yellow lines: hydrogen (H) bonds. R799 points toward the protein core where it is engaged in H‐bonds. The p.R799C mutation is predicted to abolish H‐bond formation, which may destabilize the overall protein structure. The p.T851M mutation alters a conserved P‐F/Y‐S/T motif that is required for the folding of the N‐terminal domain in 4b6z. The larger and more hydrophobic methionine could impair stability of the immediate environment and thus interfere with protein folding. R910 resides in an outwardly projecting loop. Loss of R910 H‐bonds due to the p.R910C mutation may destabilize the loop region. The p.R918W substitution affects a loop containing residues (H920, E923) coordinating the zinc ion in the active site. The bulkier tryptophan may cause steric clashes (red), disrupting the shape of the active site. The p.H990L substitution affects a residue that contributes to a network of H‐bonds, possibly important for maintaining the structure of the close‐by active site (acetate ion).
Low levels of missense‐mutant CCP1 in HEK293 cells transfected with expression vectors for mouse wild‐type and mutant CCP1‐YFP. Residues Y686, T843, R910, and H982 align to human Y694, T851, R918, and H990. Graphs: mean ± SD, n = 3. P‐values: one‐way ANOVA, Tukey post‐test.
Impaired deglutamylase activity of overexpressed CCP1 missense mutants in HEK293 cells. Inactive: catalytically dead p.H912S + E915Q13. Δ2‐tub: Δ2‐α‐tubulin generated through CCP1‐mediated removal of the gene‐encoded C‐terminal glutamate residue from α‐tubulin.
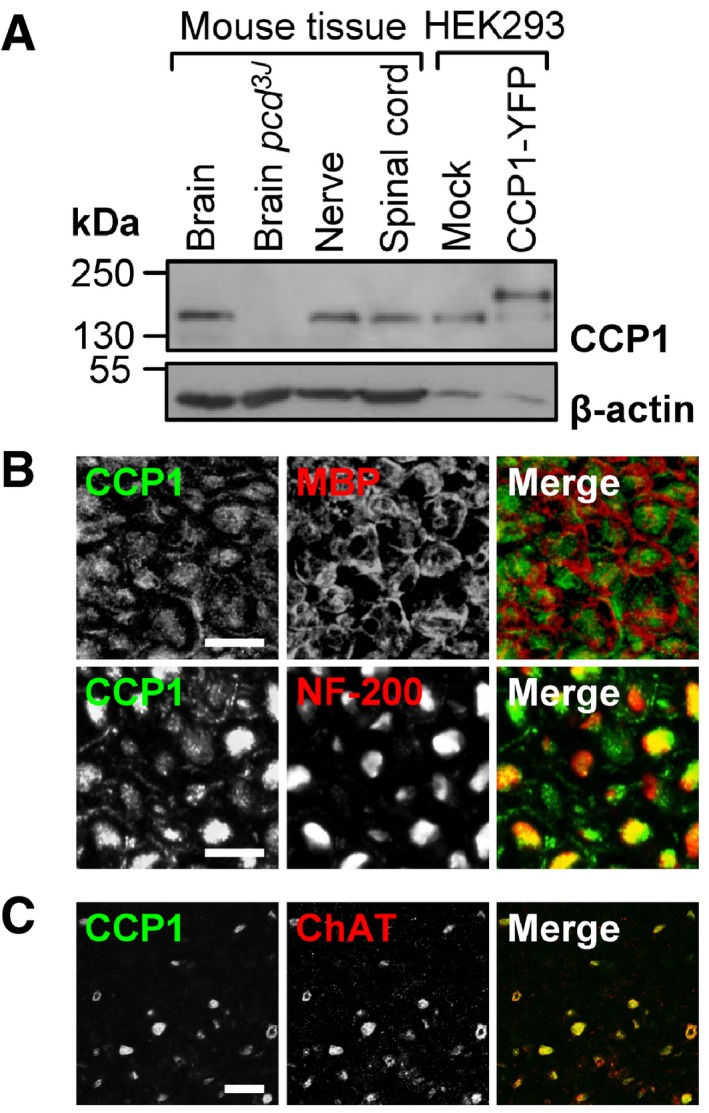
Detection of CCP1 in protein extracts from brain, spinal cord, and peripheral nerve. Specificity of the anti‐CCP1 antibody was verified by no signal in pcd mouse brain and an extra band in HEK293 cells transfected with a CCP1‐YFP fusion protein.
Association of CCP1 with myelinated axons (neurofilament (NF‐200)‐positive) but not compact myelin (myelin basic protein (MBP)‐positive) in peripheral nerve. Cross sections, scale bar, 10 μm.
Presence of CCP1 in motor neurons (choline acetyltransferase (ChAT)‐positive) in the ventral horn of the lumbar spinal cord. Cross sections, scale bar, 50 μm.
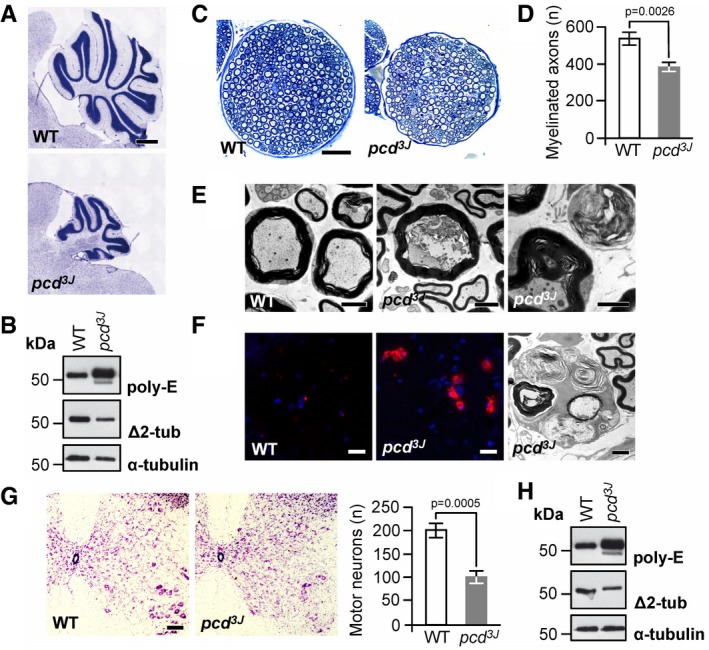
Cerebellar atrophy in pcd mice. Nissl staining of mid‐sagittal sections of cerebella at P150, displaying massively reduced size of the cerebellar vermis in pcd mice relative to wild type (WT). Scale bar, 500 μm.
Excess tubulin polyglutamylation (poly‐E) and decreased Δ2‐tubulin (Δ2‐tub) generation in pcd cerebellum. WT: wild type.
Reduction in nerve diameter and numbers of myelinated fibers in pcd mice. Cross sections of femoral quadriceps nerves, toluidine blue staining, scale bar, 50 μm.
Quantification of myelinated axons in the femoral quadriceps nerves of WT and pcd mice. Graphs: mean ± SD, n = 3. P‐value: Student's unpaired two‐tailed t‐test.
Disorganized axoplasm (middle), reduced axon diameter with preserved myelin profile and contorted myelin devoid of an axon (right). Left: normal myelinated axons (WT mouse). Cross sections of femoral quadriceps nerves, electron microscopy, scale bar, 2 μm.
Activated endoneurial macrophages in pcd mice. CD68 immunostaining (macrophage marker, left and middle image) and electron microscopy (rounded macrophages containing axonal fragments and myelin debris, right image). Scale bars, 20 μm (left and middle image), 2 μm (right image).
Reduction in the number of motor neurons (large cells in the lower right corners) in ventral horns of pcd spinal cord. Transverse sections, Nissl staining, scale bar, 100 μm. Graphs: mean ± SD, n = 3. P‐values: Student's unpaired two‐tailed t‐test.
Excess tubulin polyglutamylation (poly‐E) and decreased Δ2‐tubulin (Δ2‐tub) generation in pcd spinal cord. WT: wild type.
Comment in
-
Neurodegenerative polyglutamylation.Nat Rev Mol Cell Biol. 2019 Jan;20(1):1. doi: 10.1038/s41580-018-0083-1. Nat Rev Mol Cell Biol. 2019. PMID: 30443035 No abstract available.
-
Microtubules and Neurodegeneration: The Tubulin Code Sets the Rules of the Road.Curr Biol. 2019 Jan 7;29(1):R28-R30. doi: 10.1016/j.cub.2018.11.031. Curr Biol. 2019. PMID: 30620913
References
-
- Abercrombie M (1946) Estimation of nuclear population from microtome sections. Anat Rec 94: 239–247 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
