

Subtype-specific regulatory network rewiring in acute myeloid leukemia
- PMID: 30420649
- PMCID: PMC6330064
- DOI: 10.1038/s41588-018-0270-1
Subtype-specific regulatory network rewiring in acute myeloid leukemia
Abstract
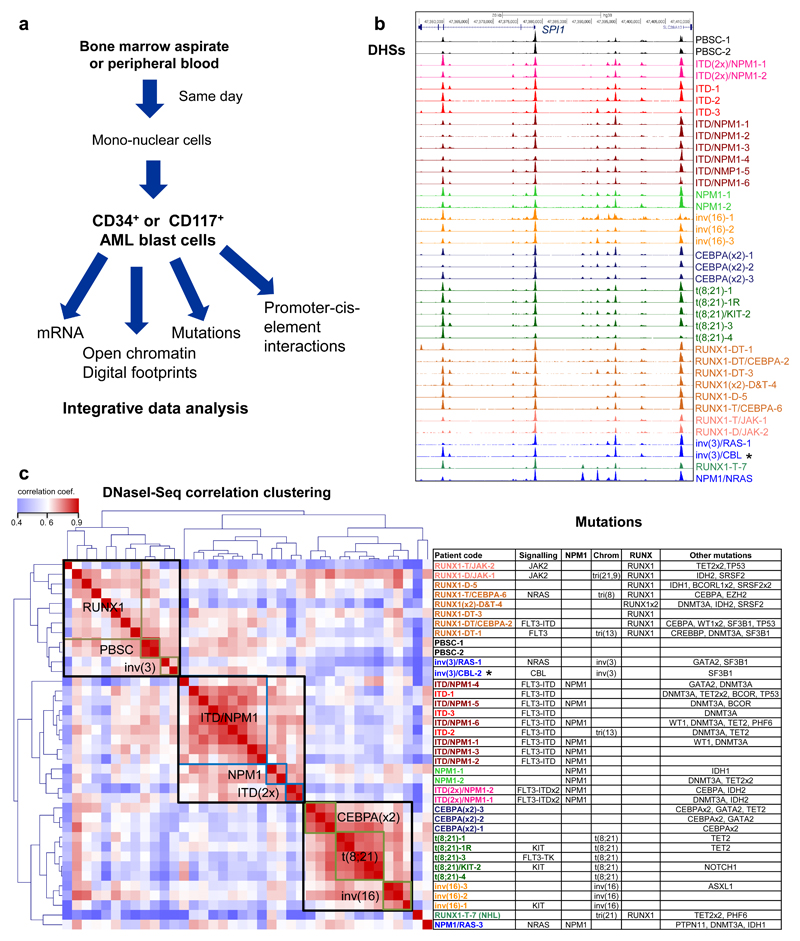
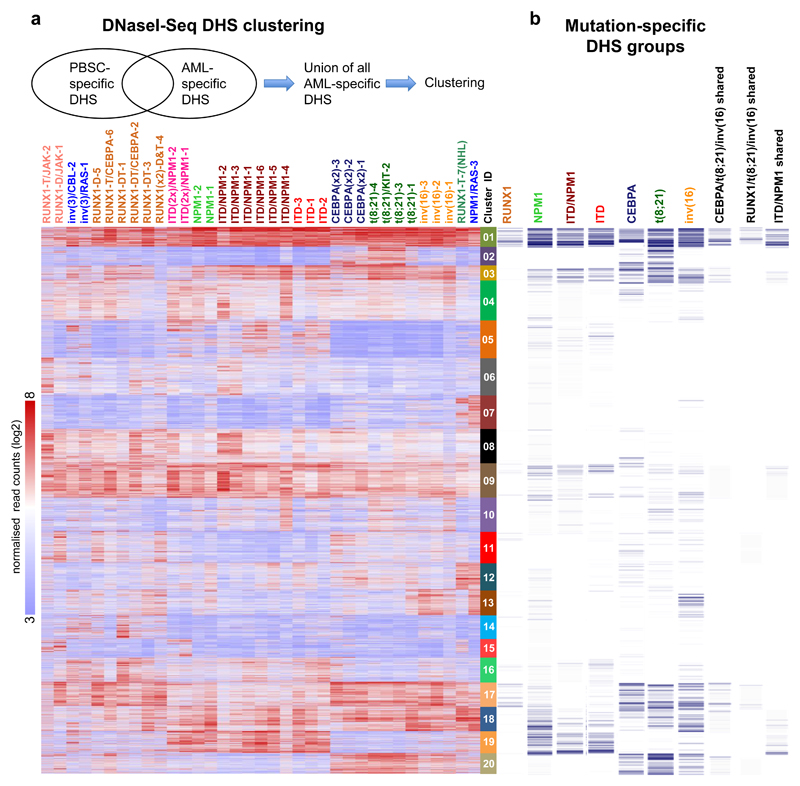
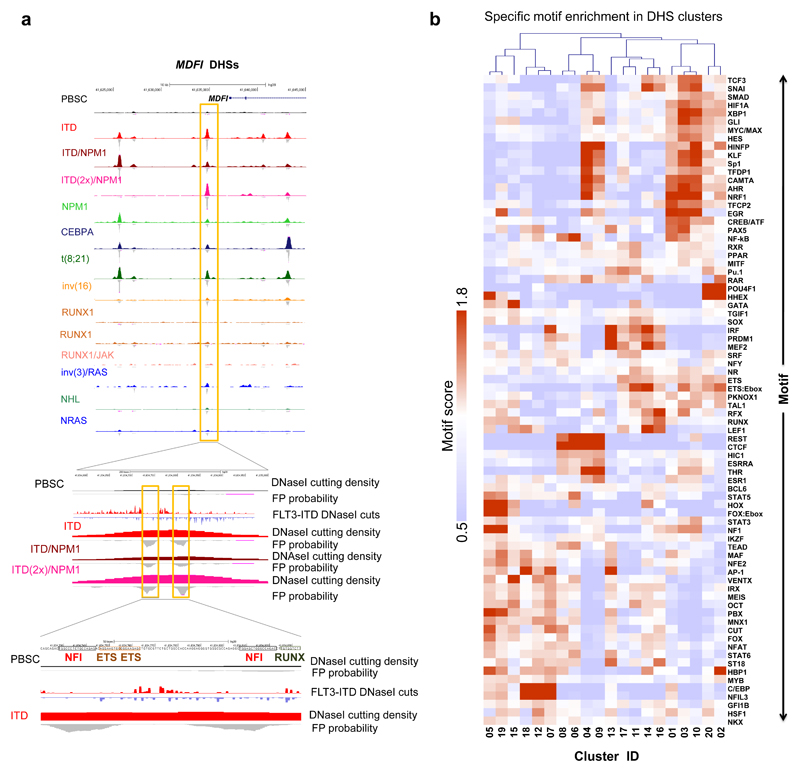
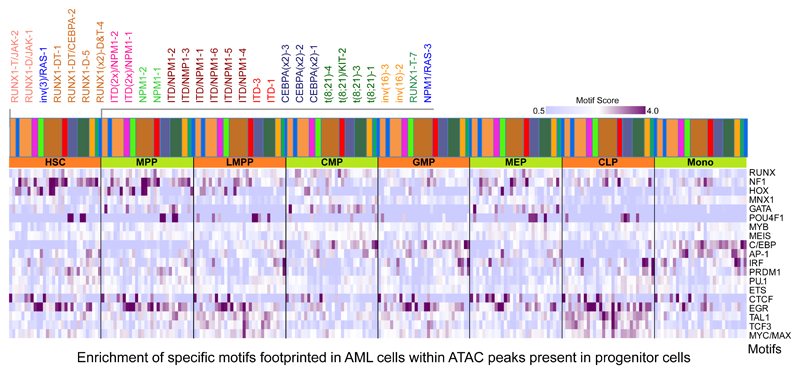
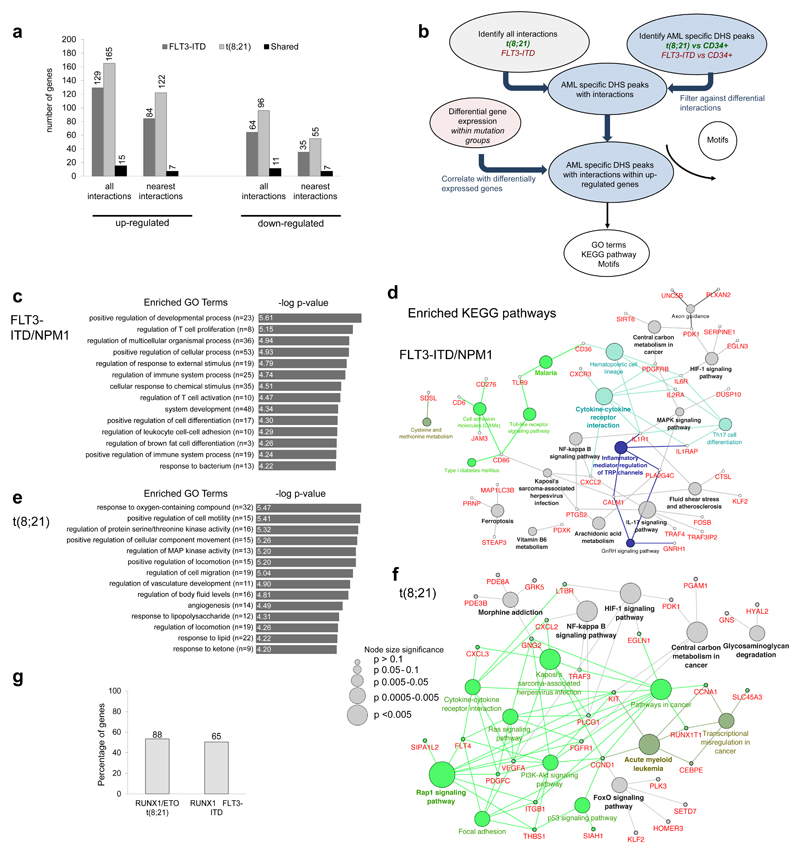
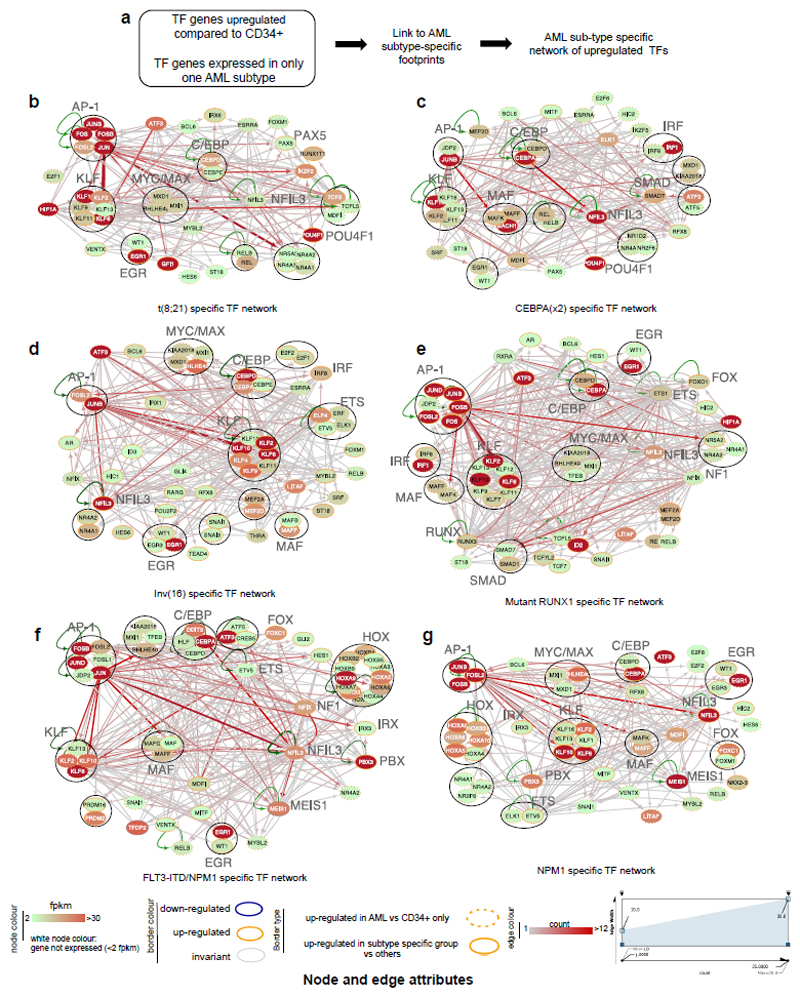
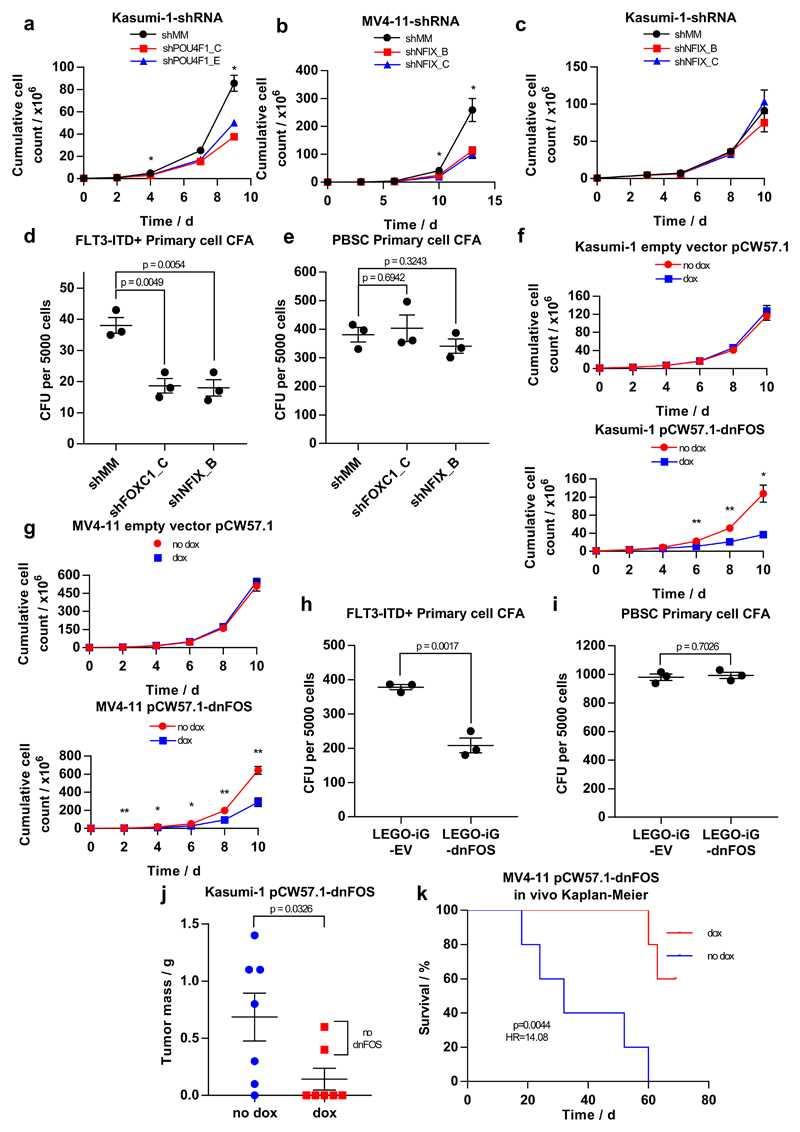
Acute myeloid leukemia (AML) is a heterogeneous disease caused by a variety of alterations in transcription factors, epigenetic regulators and signaling molecules. To determine how different mutant regulators establish AML subtype-specific transcriptional networks, we performed a comprehensive global analysis of cis-regulatory element activity and interaction, transcription factor occupancy and gene expression patterns in purified leukemic blast cells. Here, we focused on specific subgroups of subjects carrying mutations in genes encoding transcription factors (RUNX1, CEBPα), signaling molecules (FTL3-ITD, RAS) and the nuclear protein NPM1). Integrated analysis of these data demonstrates that each mutant regulator establishes a specific transcriptional and signaling network unrelated to that seen in normal cells, sustaining the expression of unique sets of genes required for AML growth and maintenance.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Regulatory networks in AML.Nat Rev Cancer. 2019 Jan;19(1):6-7. doi: 10.1038/s41568-018-0092-6. Nat Rev Cancer. 2019. PMID: 30523341 No abstract available.
-
Rewiring of the Transcription Factor Network in Acute Myeloid Leukemia.Cancer Inform. 2019 Jun 25;18:1176935119859863. doi: 10.1177/1176935119859863. eCollection 2019. Cancer Inform. 2019. PMID: 31263370 Free PMC article.
References
-
- Bonifer C, Cockerill PN. Chromatin Structure Profiling Identifies Crucial Regulators of Tumor Maintenance. Trends Cancer. 2015;1:157–160. - PubMed
-
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nature reviews. Immunology. 2007;7:105–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
