

A204E mutation in Nav1.4 DIS3 exerts gain- and loss-of-function effects that lead to periodic paralysis combining hyper- with hypo-kalaemic signs
- PMID: 30420713
- PMCID: PMC6232142
- DOI: 10.1038/s41598-018-34750-8
A204E mutation in Nav1.4 DIS3 exerts gain- and loss-of-function effects that lead to periodic paralysis combining hyper- with hypo-kalaemic signs
Abstract
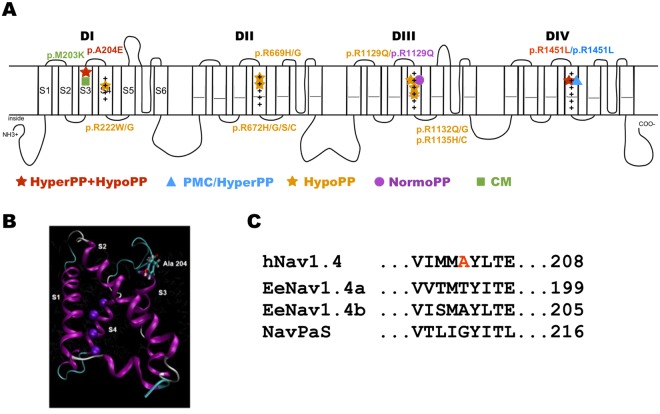
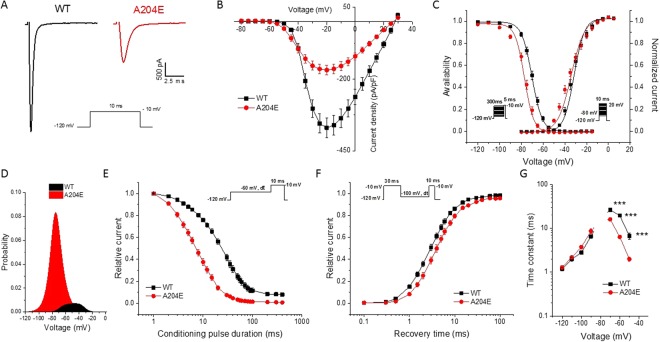
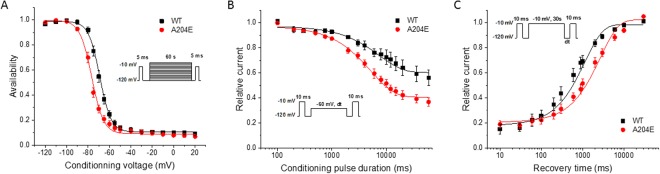
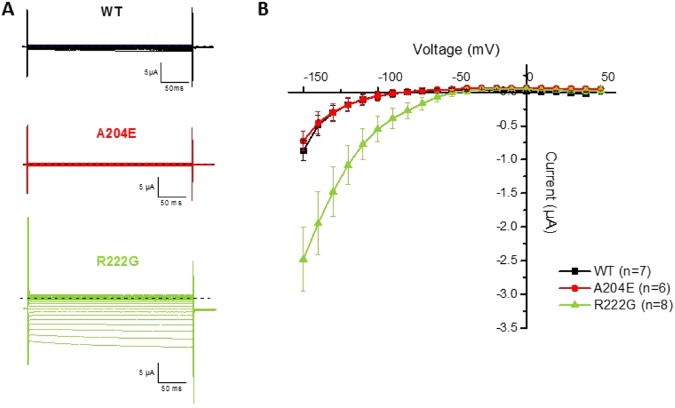
Periodic paralyses (PP) are characterized by episodic muscle weakness and are classified into the distinct hyperkalaemic (hyperPP) and hypokalaemic (hypoPP) forms. The dominantly-inherited form of hyperPP is caused by overactivity of Nav1.4 - the skeletal muscle voltage-gated sodium channel. Familial hypoPP results from a leaking gating pore current induced by dominant mutations in Nav1.4 or Cav1.1, the skeletal muscle voltage-gated calcium channel. Here, we report an individual with clinical signs of hyperPP and hypokalaemic episodes of muscle paralysis who was heterozygous for the novel p.Ala204Glu (A204E) substitution located in one region of Nav1.4 poor in disease-related variations. A204E induced a significant decrease of sodium current density, increased the window current, enhanced fast and slow inactivation of Nav1.4, and did not cause gating pore current in functional analyses. Interestingly, the negative impact of A204E on Nav1.4 activation was strengthened in low concentration of extracellular K+. Our data prove the existence of a phenotype combining signs of hyperPP and hypoPP due to dominant Nav1.4 mutations. The hyperPP component would result from gain-of-function effects on Nav1.4 and the hypokalemic episodes of paralysis from loss-of-function effects strengthened by low K+. Our data argue for a non-negligible role of Nav1.4 loss-of-function in familial hypoPP.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Hypokalaemic periodic paralysis and myotonia in a patient with homozygous mutation p.R1451L in NaV1.4.Sci Rep. 2018 Jun 26;8(1):9714. doi: 10.1038/s41598-018-27822-2. Sci Rep. 2018. PMID: 29946067 Free PMC article.
-
Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation.Sci Rep. 2019 Nov 26;9(1):17560. doi: 10.1038/s41598-019-54041-0. Sci Rep. 2019. PMID: 31772215 Free PMC article.
-
Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current.Proc Natl Acad Sci U S A. 2000 Aug 15;97(17):9549-54. doi: 10.1073/pnas.97.17.9549. Proc Natl Acad Sci U S A. 2000. PMID: 10944223 Free PMC article.
-
Muscle channelopathies: does the predicted channel gating pore offer new treatment insights for hypokalaemic periodic paralysis?J Physiol. 2010 Jun 1;588(Pt 11):1879-86. doi: 10.1113/jphysiol.2009.186627. Epub 2010 Feb 1. J Physiol. 2010. PMID: 20123788 Free PMC article. Review.
-
Periodic paralysis: understanding channelopathies.Curr Neurol Neurosci Rep. 2002 Jan;2(1):61-9. doi: 10.1007/s11910-002-0055-9. Curr Neurol Neurosci Rep. 2002. PMID: 11898585 Review.
Cited by
-
Inherited Neuromuscular Disorders: Which Role for Serum Biomarkers?Brain Sci. 2021 Mar 21;11(3):398. doi: 10.3390/brainsci11030398. Brain Sci. 2021. PMID: 33801069 Free PMC article. Review.
-
New Challenges Resulting From the Loss of Function of Nav1.4 in Neuromuscular Diseases.Front Pharmacol. 2021 Oct 4;12:751095. doi: 10.3389/fphar.2021.751095. eCollection 2021. Front Pharmacol. 2021. PMID: 34671263 Free PMC article. Review.
-
Hypokalaemic periodic paralysis with a charge-retaining substitution in the voltage sensor.Brain Commun. 2020 Jul 16;2(2):fcaa103. doi: 10.1093/braincomms/fcaa103. eCollection 2020. Brain Commun. 2020. PMID: 33005891 Free PMC article.
-
Molecular genetics of skeletal muscle channelopathies.J Hum Genet. 2025 Aug 6. doi: 10.1038/s10038-025-01370-w. Online ahead of print. J Hum Genet. 2025. PMID: 40770230 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
