

Cardiac CaV1.2 channels require β subunits for β-adrenergic-mediated modulation but not trafficking
- PMID: 30422117
- PMCID: PMC6355231
- DOI: 10.1172/JCI123878
Cardiac CaV1.2 channels require β subunits for β-adrenergic-mediated modulation but not trafficking
Abstract
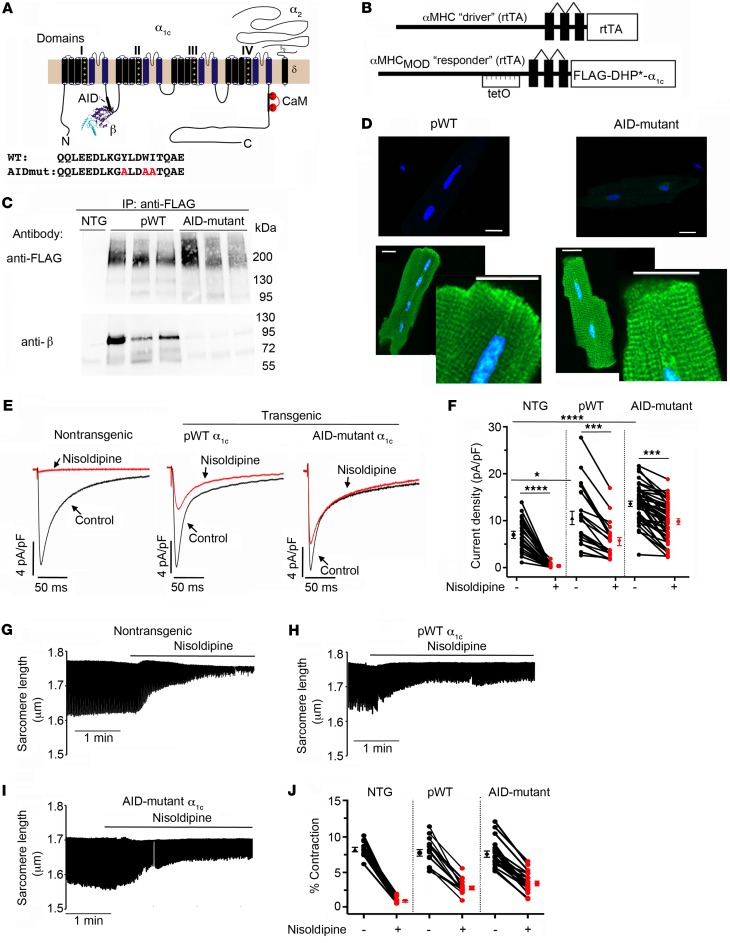
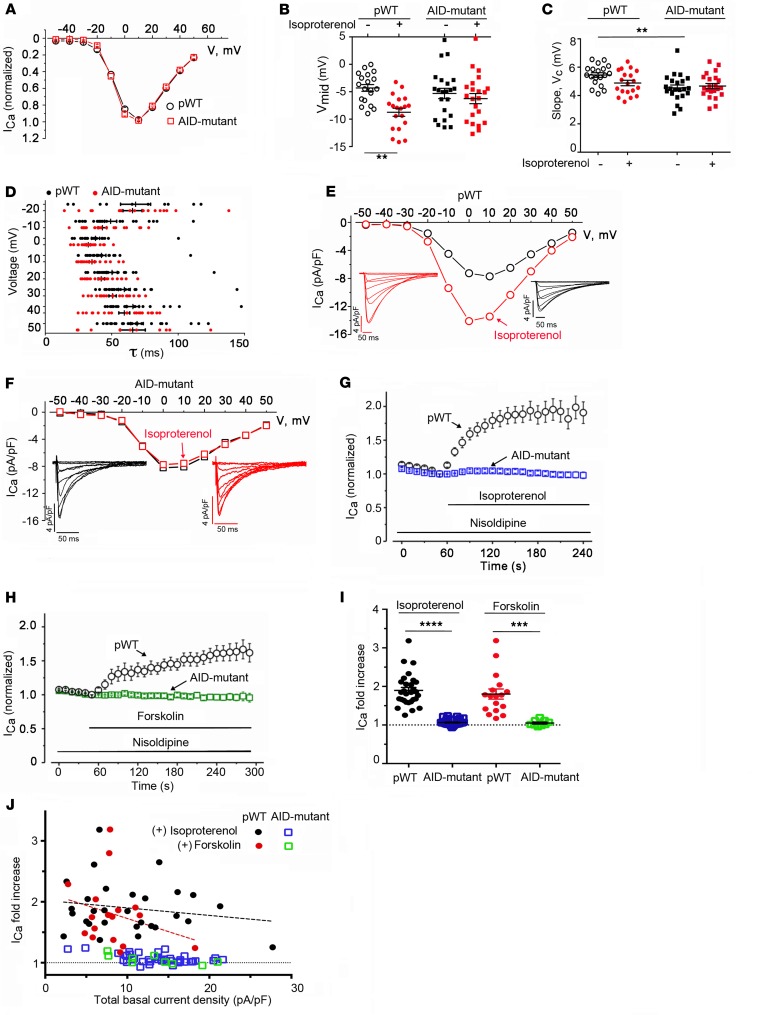
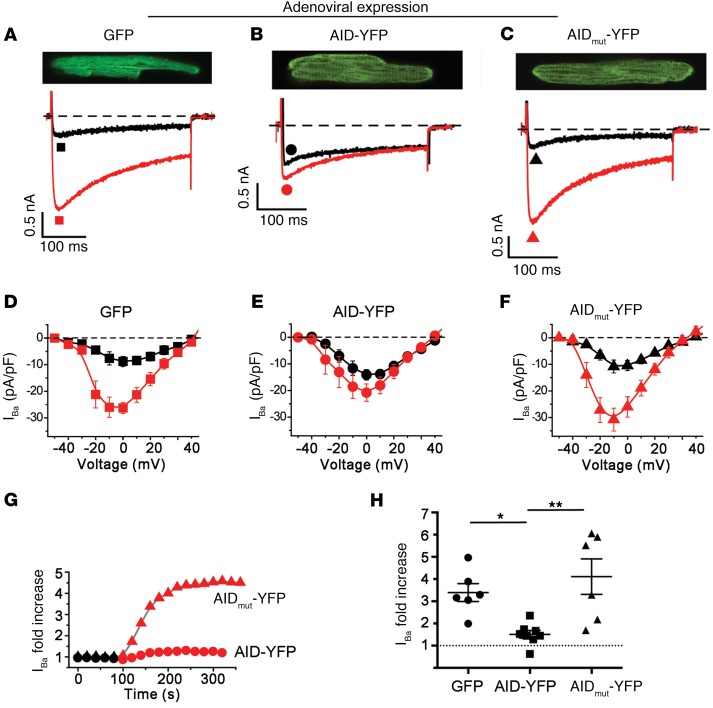
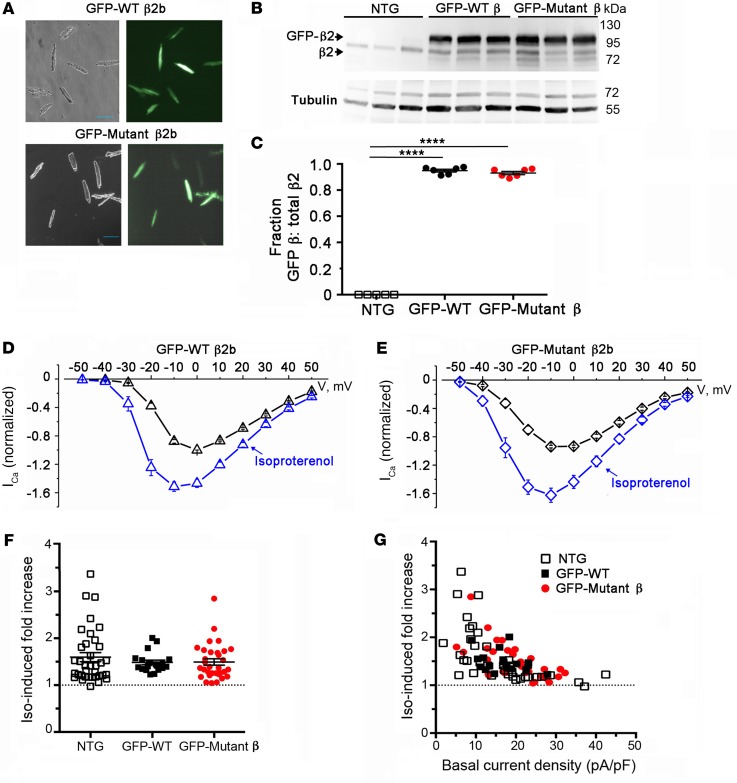
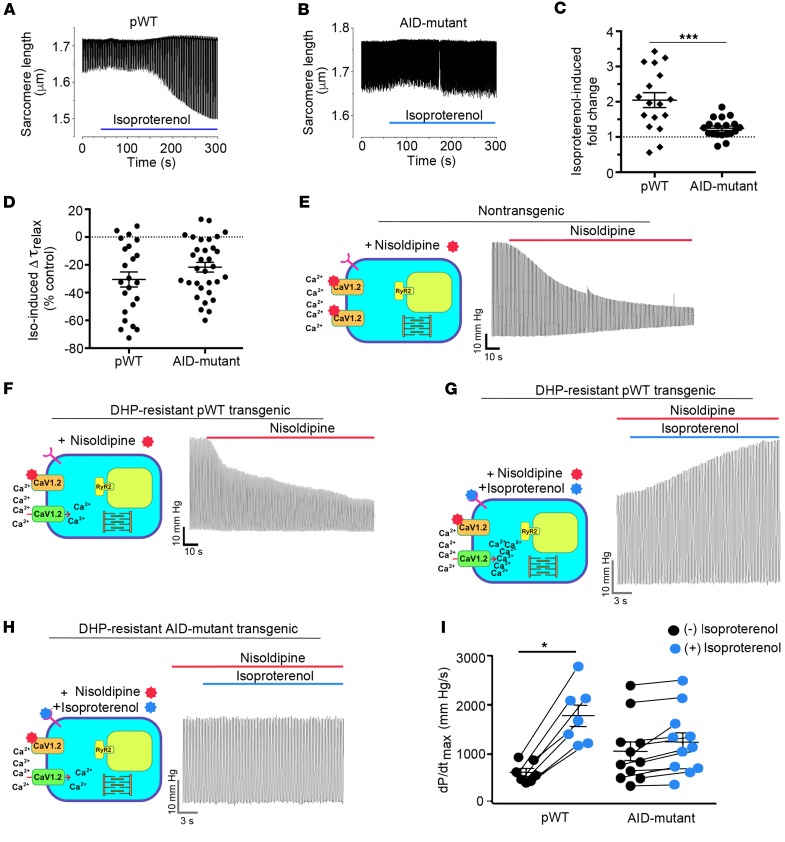
Ca2+ channel β-subunit interactions with pore-forming α-subunits are long-thought to be obligatory for channel trafficking to the cell surface and for tuning of basal biophysical properties in many tissues. Unexpectedly, we demonstrate that transgenic expression of mutant α1C subunits lacking capacity to bind CaVβ can traffic to the sarcolemma in adult cardiomyocytes in vivo and sustain normal excitation-contraction coupling. However, these β-less Ca2+ channels cannot be stimulated by β-adrenergic pathway agonists, and thus adrenergic augmentation of contractility is markedly impaired in isolated cardiomyocytes and in hearts. Similarly, viral-mediated expression of a β-subunit-sequestering peptide sharply curtailed β-adrenergic stimulation of WT Ca2+ channels, identifying an approach to specifically modulate β-adrenergic regulation of cardiac contractility. Our data demonstrate that β subunits are required for β-adrenergic regulation of CaV1.2 channels and positive inotropy in the heart, but are dispensable for CaV1.2 trafficking to the adult cardiomyocyte cell surface, and for basal function and excitation-contraction coupling.
Keywords: Calcium; Calcium channels; Cardiology; Excitation contraction coupling; Muscle Biology.
Conflict of interest statement
Figures
Comment in
-
The L-type calcium channel current modulation mechanism: the plot thickens and fogs.J Clin Invest. 2019 Feb 1;129(2):496-498. doi: 10.1172/JCI125958. Epub 2019 Jan 7. J Clin Invest. 2019. PMID: 30614816 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
