

SPhyR: tumor phylogeny estimation from single-cell sequencing data under loss and error
- PMID: 30423070
- PMCID: PMC6153375
- DOI: 10.1093/bioinformatics/bty589
SPhyR: tumor phylogeny estimation from single-cell sequencing data under loss and error
Abstract
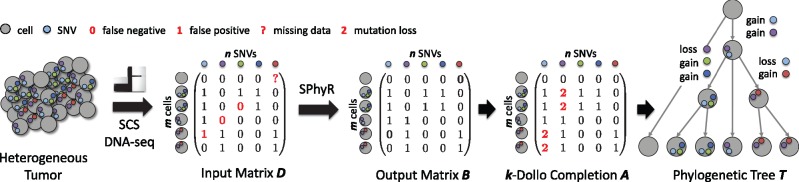
Motivation: Cancer is characterized by intra-tumor heterogeneity, the presence of distinct cell populations with distinct complements of somatic mutations, which include single-nucleotide variants (SNVs) and copy-number aberrations (CNAs). Single-cell sequencing technology enables one to study these cell populations at single-cell resolution. Phylogeny estimation algorithms that employ appropriate evolutionary models are key to understanding the evolutionary mechanisms behind intra-tumor heterogeneity.
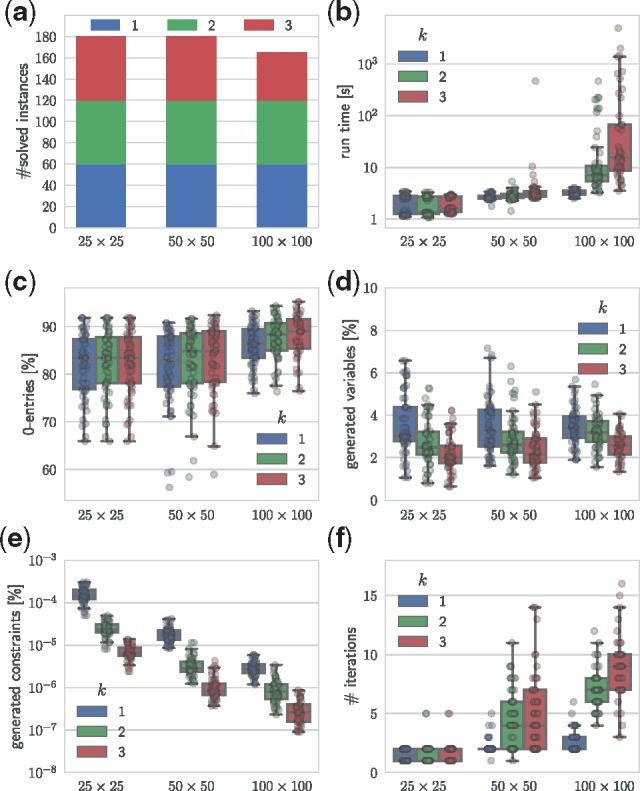
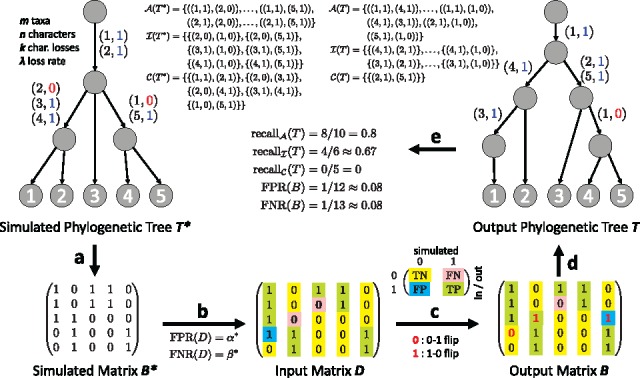
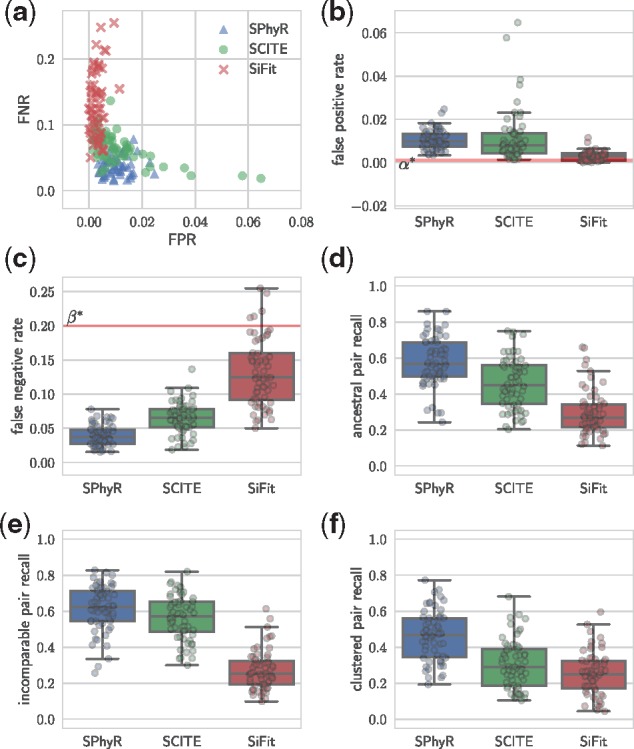
Results: We introduce Single-cell Phylogeny Reconstruction (SPhyR), a method for tumor phylogeny estimation from single-cell sequencing data. In light of frequent loss of SNVs due to CNAs in cancer, SPhyR employs the k-Dollo evolutionary model, where a mutation can only be gained once but lost k times. Underlying SPhyR is a novel combinatorial characterization of solutions as constrained integer matrix completions, based on a connection to the cladistic multi-state perfect phylogeny problem. SPhyR outperforms existing methods on simulated data and on a metastatic colorectal cancer.
Availability and implementation: SPhyR is available on https://github.com/elkebir-group/SPhyR.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Agarwala R., Fernández-Baca D. (1994) A polynomial-time algorithm for the perfect phylogeny problem when the number of character states is fixed. SIAM J. Comput., 23, 1216–1224.
-
- Bodlaender H.L., et al. (1992) Two strikes against perfect phylogeny. In: Kuich W. (ed.) Automata, Languages and Programming. ICALP 1992. Lecture Notes in Computer Science. Springer, Berlin, Heidelberg: Vol 623.
-
- Bonizzoni P., et al. (2012) The binary perfect phylogeny with persistent characters. Theor. Comput. Sci., 454, 51–63.
-
- Bonizzoni P., et al. (2017a) A colored graph approach to perfect phylogeny with persistent characters. Theor. Comput. Sci., 658, 60–73.
-
- Bonizzoni P., et al. (2017b) Beyond perfect phylogeny: multisample phylogeny reconstruction via ilp. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics ,ACM-BCB ‘17, ACM, New York, NY, USA. pp. 1–10.
