

fastp: an ultra-fast all-in-one FASTQ preprocessor
- PMID: 30423086
- PMCID: PMC6129281
- DOI: 10.1093/bioinformatics/bty560
fastp: an ultra-fast all-in-one FASTQ preprocessor
Abstract
Motivation: Quality control and preprocessing of FASTQ files are essential to providing clean data for downstream analysis. Traditionally, a different tool is used for each operation, such as quality control, adapter trimming and quality filtering. These tools are often insufficiently fast as most are developed using high-level programming languages (e.g. Python and Java) and provide limited multi-threading support. Reading and loading data multiple times also renders preprocessing slow and I/O inefficient.
Results: We developed fastp as an ultra-fast FASTQ preprocessor with useful quality control and data-filtering features. It can perform quality control, adapter trimming, quality filtering, per-read quality pruning and many other operations with a single scan of the FASTQ data. This tool is developed in C++ and has multi-threading support. Based on our evaluation, fastp is 2-5 times faster than other FASTQ preprocessing tools such as Trimmomatic or Cutadapt despite performing far more operations than similar tools.
Availability and implementation: The open-source code and corresponding instructions are available at https://github.com/OpenGene/fastp.
Figures
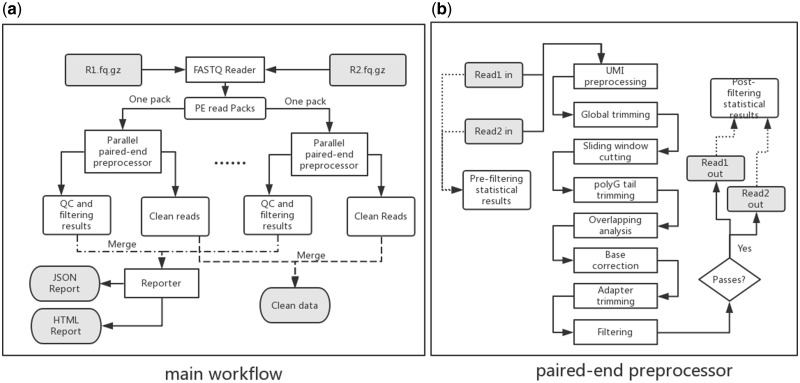
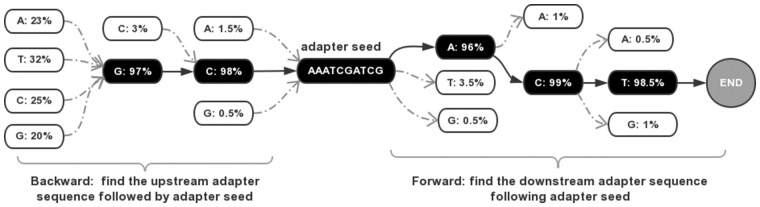
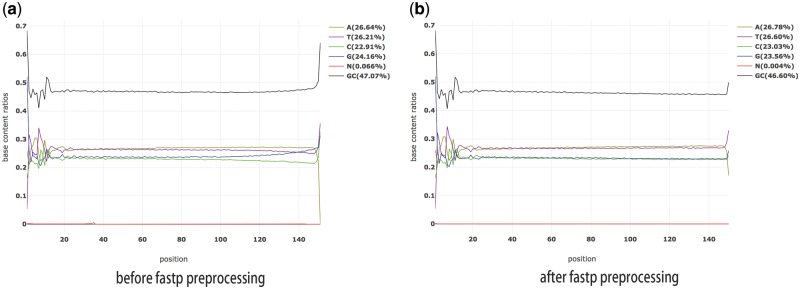
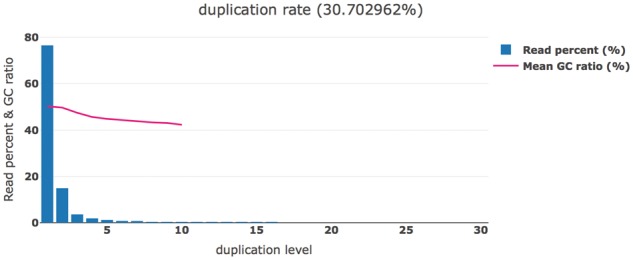
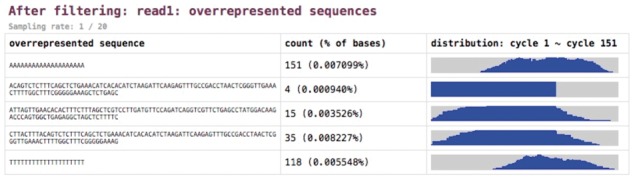
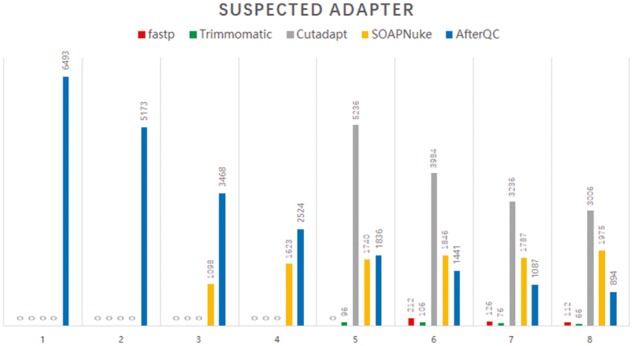
References
-
- Andrews S. (2010) A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
-
- Bianchi D.W., et al. (2015) Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA, 314, 162–169. - PubMed
-
- Brad Chapman R.K., et al. (2018) Validated, Scalable, Community Developed Variant Calling, RNA-Seq and Small RNA Analysis, https://github.com/chapmanb/bcbio-nextgen.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
