

DeepDTA: deep drug-target binding affinity prediction
- PMID: 30423097
- PMCID: PMC6129291
- DOI: 10.1093/bioinformatics/bty593
DeepDTA: deep drug-target binding affinity prediction
Abstract
Motivation: The identification of novel drug-target (DT) interactions is a substantial part of the drug discovery process. Most of the computational methods that have been proposed to predict DT interactions have focused on binary classification, where the goal is to determine whether a DT pair interacts or not. However, protein-ligand interactions assume a continuum of binding strength values, also called binding affinity and predicting this value still remains a challenge. The increase in the affinity data available in DT knowledge-bases allows the use of advanced learning techniques such as deep learning architectures in the prediction of binding affinities. In this study, we propose a deep-learning based model that uses only sequence information of both targets and drugs to predict DT interaction binding affinities. The few studies that focus on DT binding affinity prediction use either 3D structures of protein-ligand complexes or 2D features of compounds. One novel approach used in this work is the modeling of protein sequences and compound 1D representations with convolutional neural networks (CNNs).
Results: The results show that the proposed deep learning based model that uses the 1D representations of targets and drugs is an effective approach for drug target binding affinity prediction. The model in which high-level representations of a drug and a target are constructed via CNNs achieved the best Concordance Index (CI) performance in one of our larger benchmark datasets, outperforming the KronRLS algorithm and SimBoost, a state-of-the-art method for DT binding affinity prediction.
Availability and implementation: https://github.com/hkmztrk/DeepDTA.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
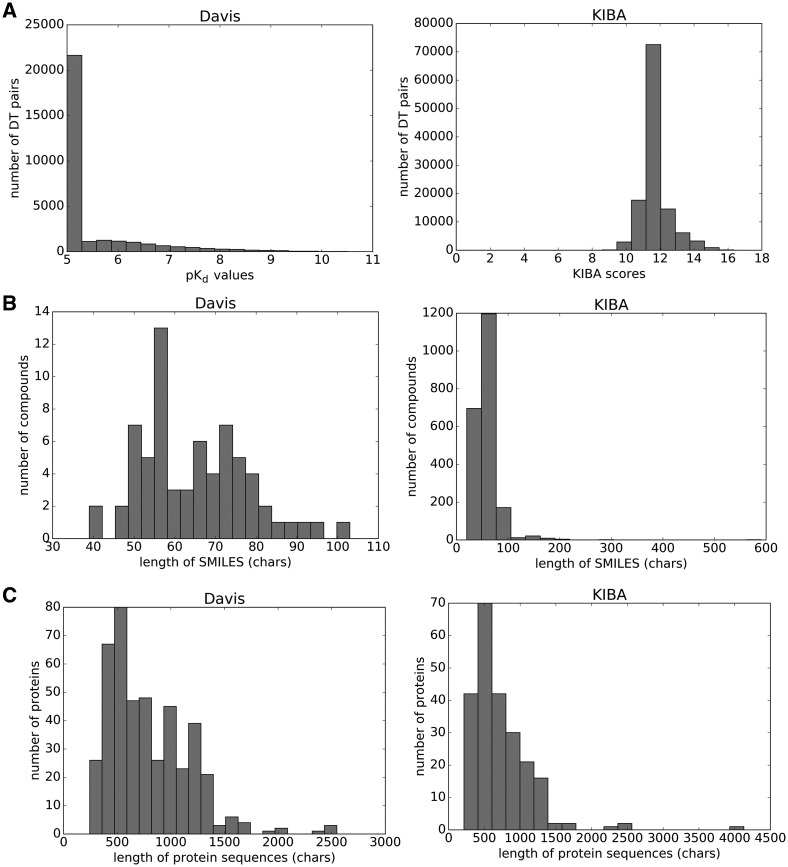
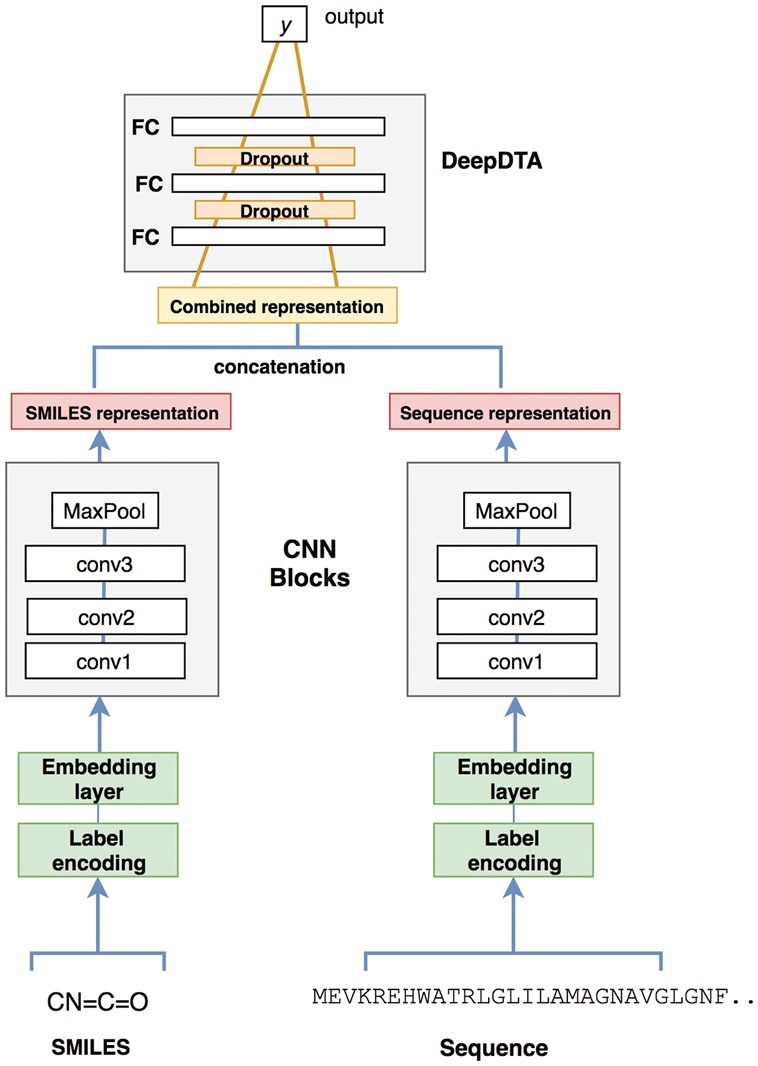
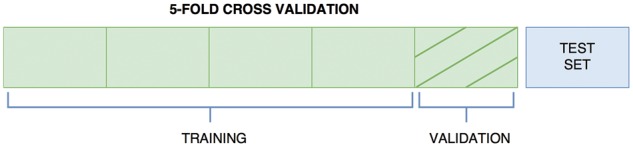
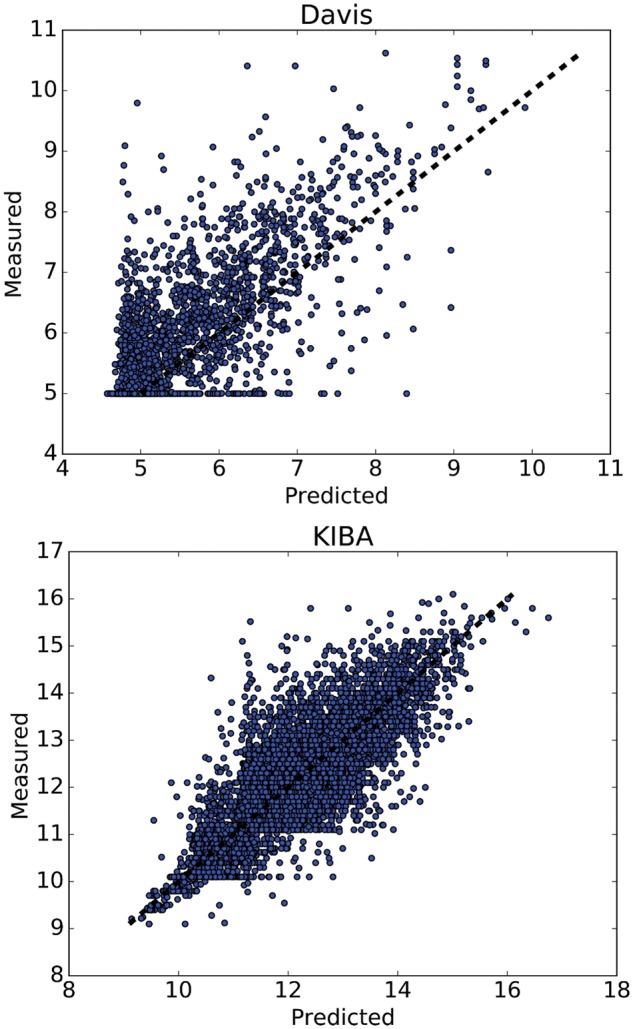
References
-
- Abadi M., et al. (2016) Tensorflow: a system for large-scale machine learning. In: OSDI, Vol. 16, pp. 265–283.
-
- Bolton E.E., et al. (2008) Pubchem: integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem., 4, 217–241.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
