

RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer
- PMID: 30423296
- PMCID: PMC6836726
- DOI: 10.1016/j.ccell.2018.10.006
RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer
Erratum in
-
RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer.Cancer Cell. 2020 Oct 12;38(4):585-590. doi: 10.1016/j.ccell.2020.09.020. Cancer Cell. 2020. PMID: 33049209 No abstract available.
Abstract
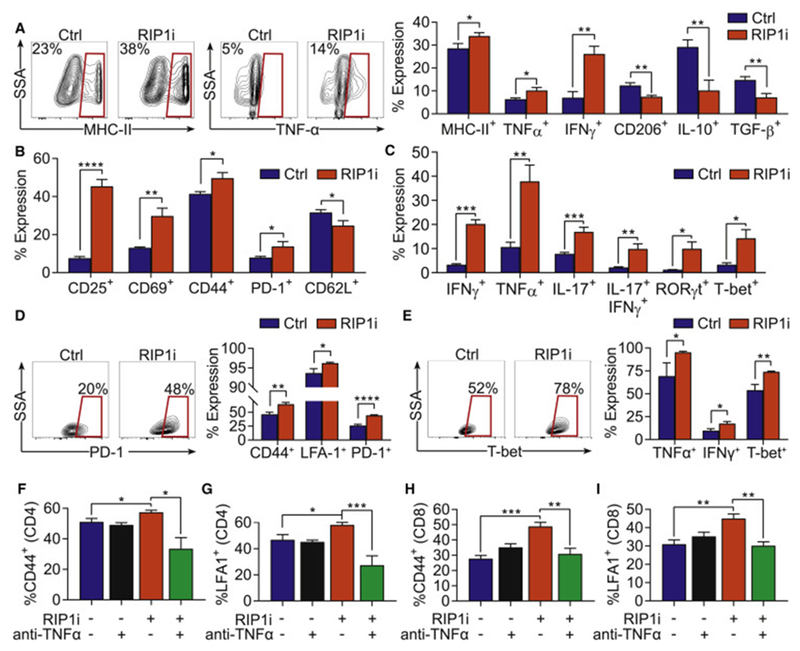
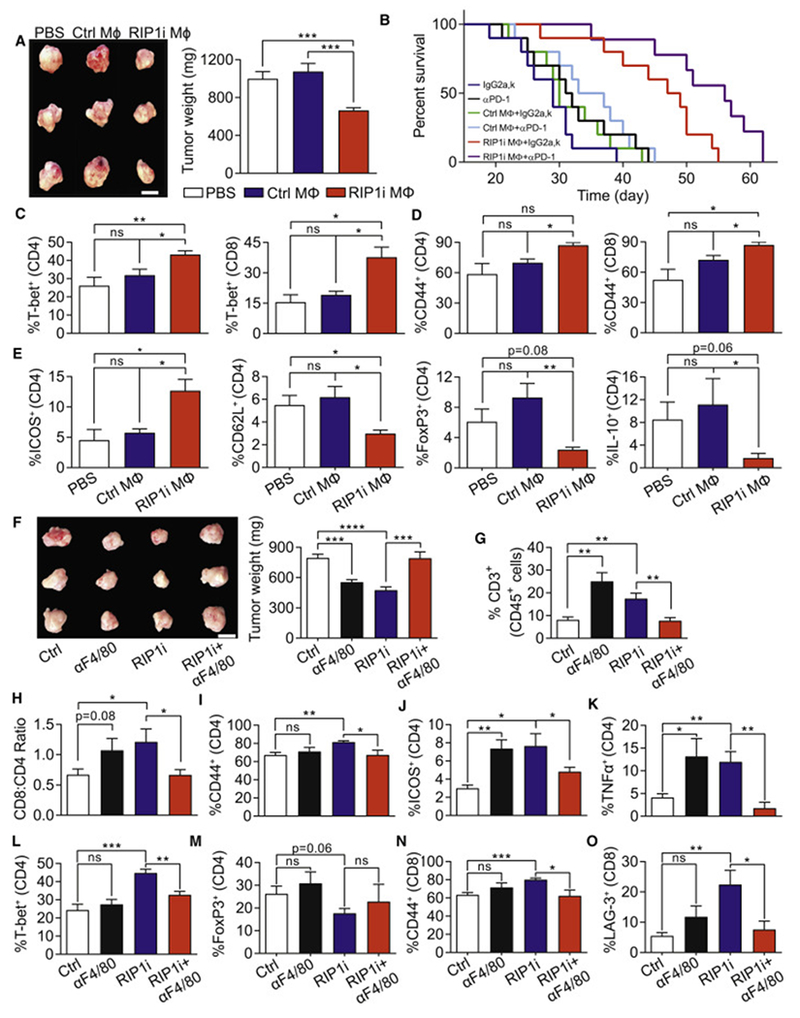
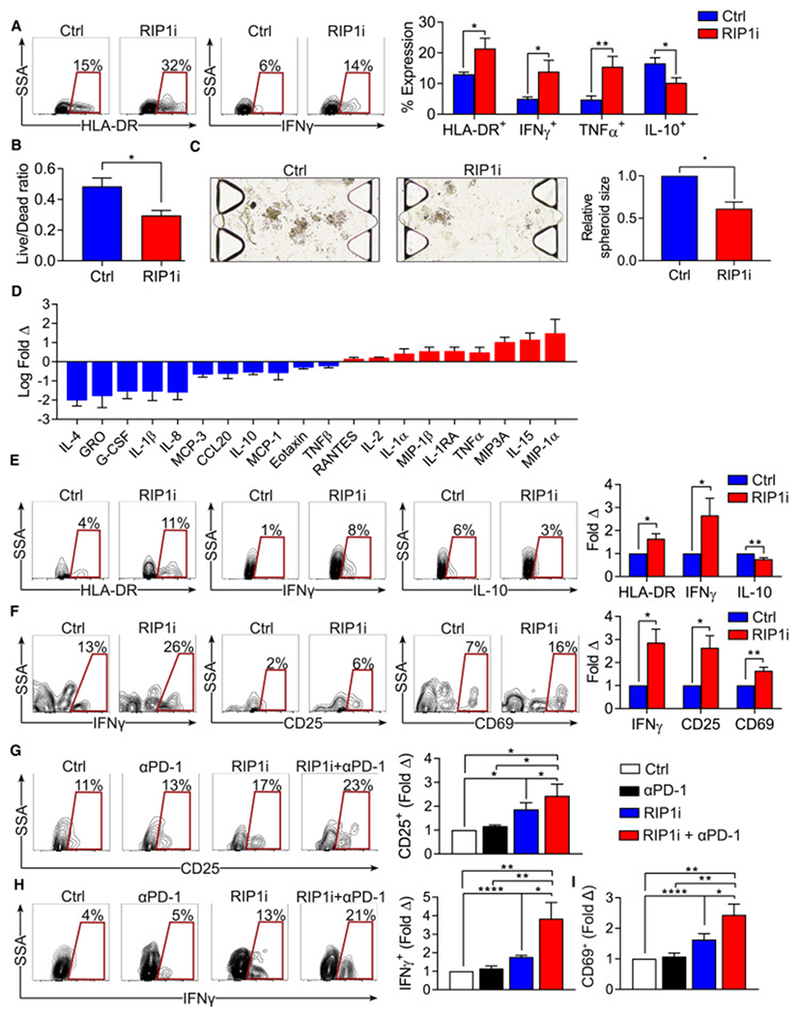
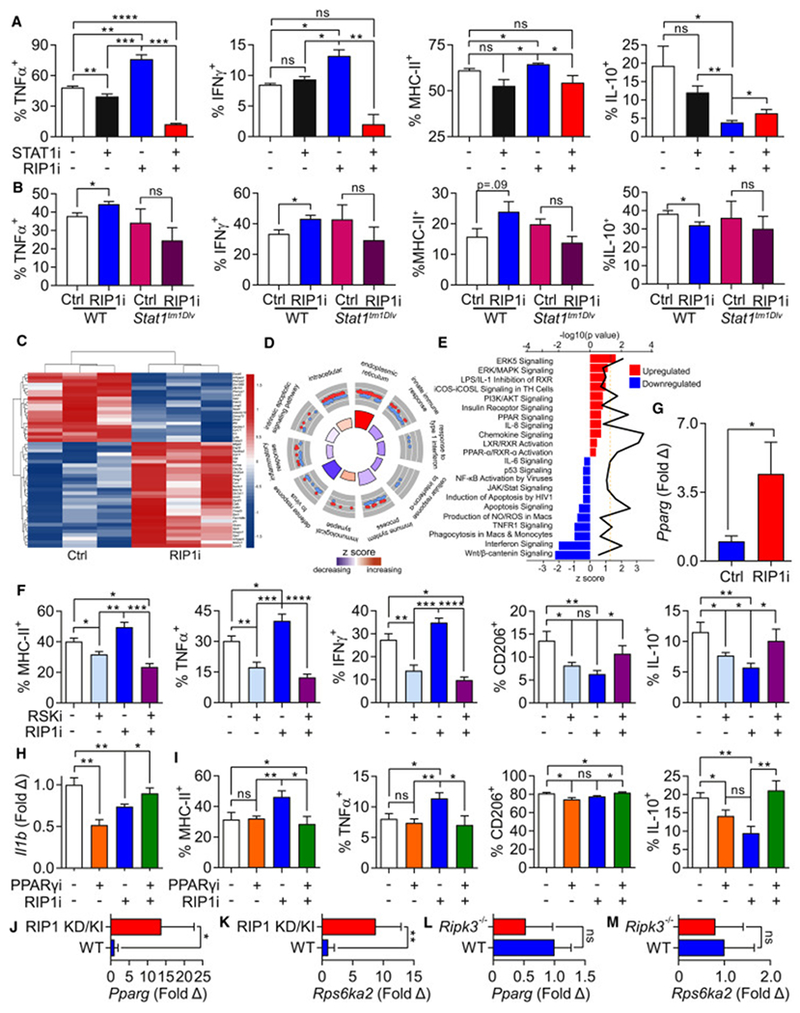
Pancreatic ductal adenocarcinoma (PDA) is characterized by immune tolerance and immunotherapeutic resistance. We discovered upregulation of receptor-interacting serine/threonine protein kinase 1 (RIP1) in tumor-associated macrophages (TAMs) in PDA. To study its role in oncogenic progression, we developed a selective small-molecule RIP1 inhibitor with high in vivo exposure. Targeting RIP1 reprogrammed TAMs toward an MHCIIhiTNFα+IFNγ+ immunogenic phenotype in a STAT1-dependent manner. RIP1 inhibition in TAMs resulted in cytotoxic T cell activation and T helper cell differentiation toward a mixed Th1/Th17 phenotype, leading to tumor immunity in mice and in organotypic models of human PDA. Targeting RIP1 synergized with PD1-and inducible co-stimulator-based immunotherapies. Tumor-promoting effects of RIP1 were independent of its co-association with RIP3. Collectively, our work describes RIP1 as a checkpoint kinase governing tumor immunity.
Keywords: Pancreatic cancer; inflammation; macrophage polarization; tumor immunity.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
JMM, AMB, MR, JL, PAH, MM, JHT, JEM, and JB are employees and shareholders of GSK.
JMM and JB are inventors on a patent application owned by GSK.
NYU School of Medicine signed a Research Contract Agreement with GSK to fund research in GM’s lab.
GM and DC received a one-time honorarium payment for giving a presentation at GSK.
GM is a co-founder and on the Scientific Advisory Board of NYBO Therapeutics.
Figures
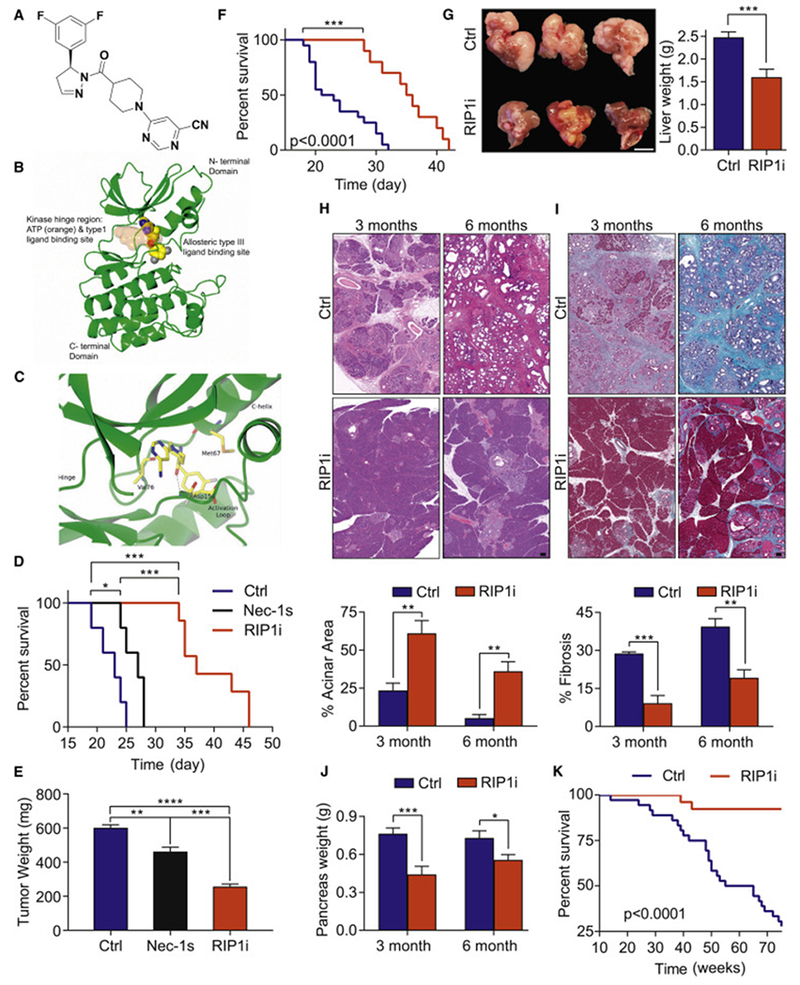
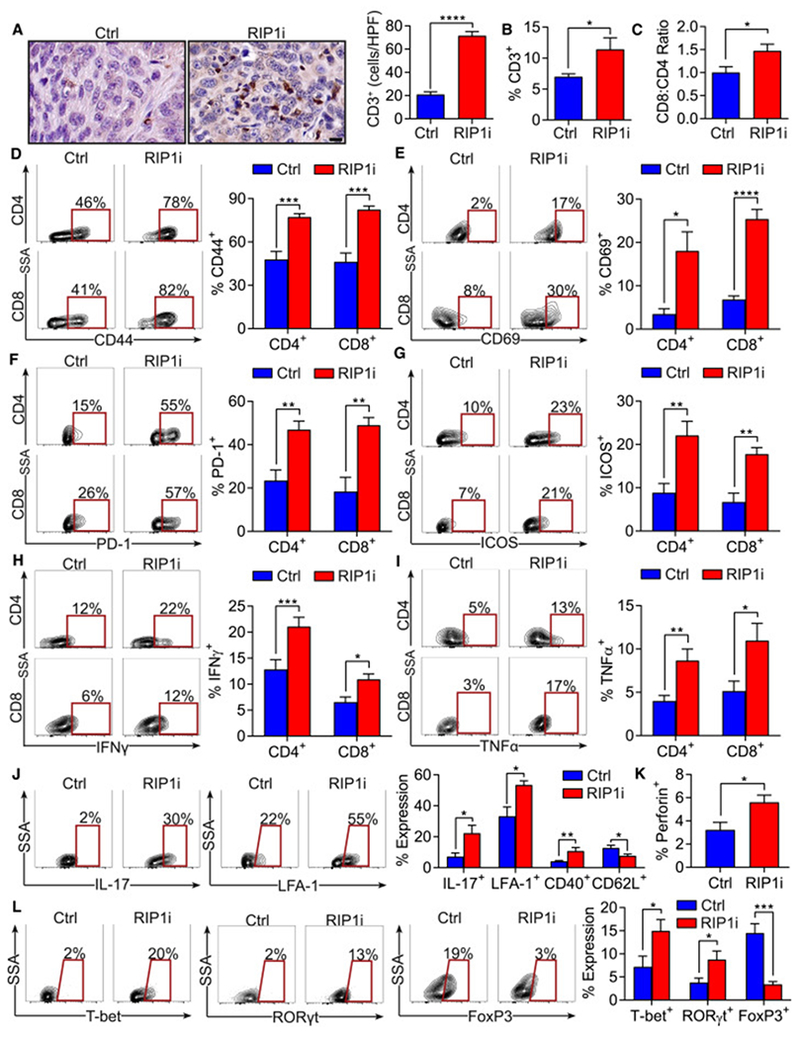
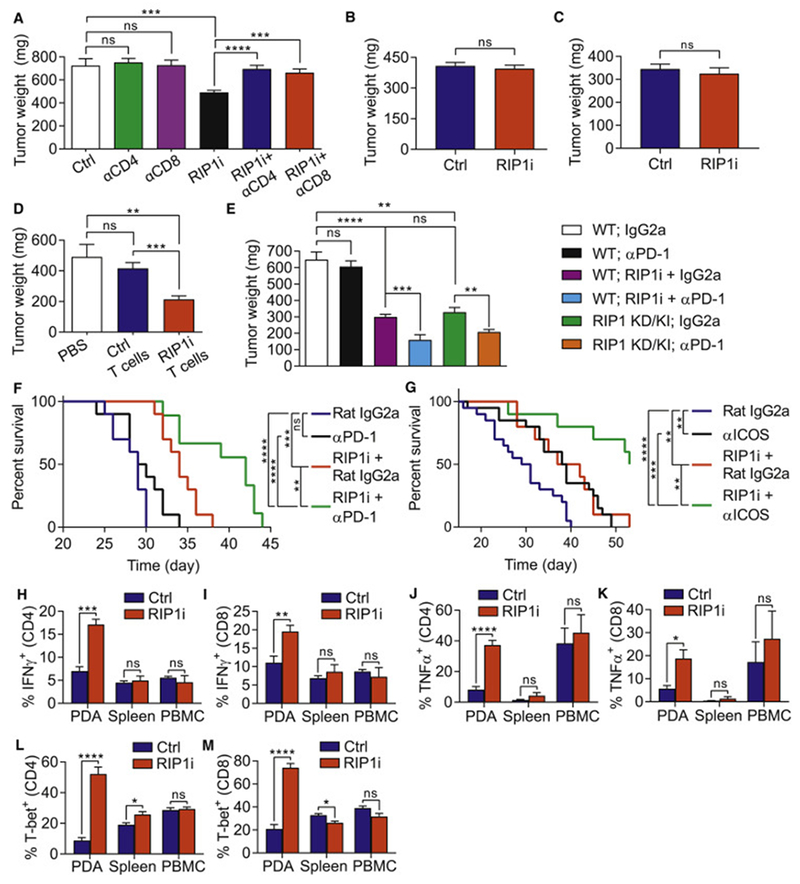
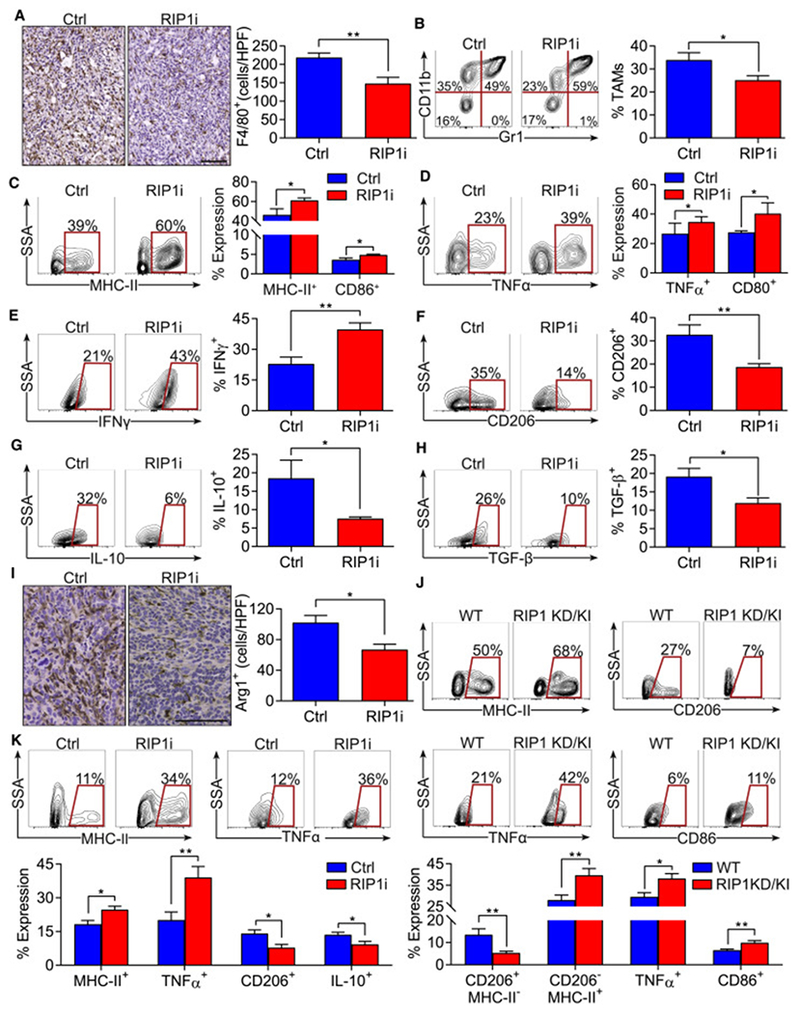
References
-
- Andren-Sandberg A, Dervenis C, and Lowenfels B (1997). Etiologic links between chronic pancreatitis and pancreatic cancer. Scand J Gastroenterol 32, 97–103. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
