

Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects
- PMID: 30424581
- PMCID: PMC6316574
- DOI: 10.3390/biomedicines6040106
Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects
Abstract
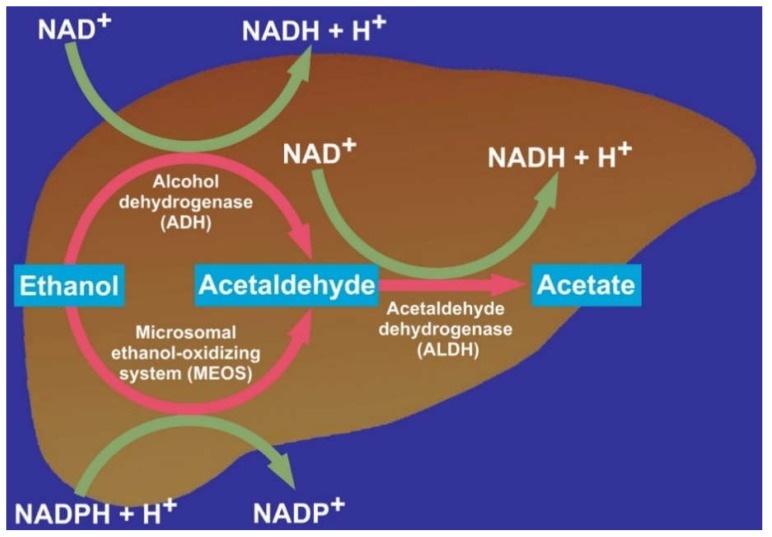
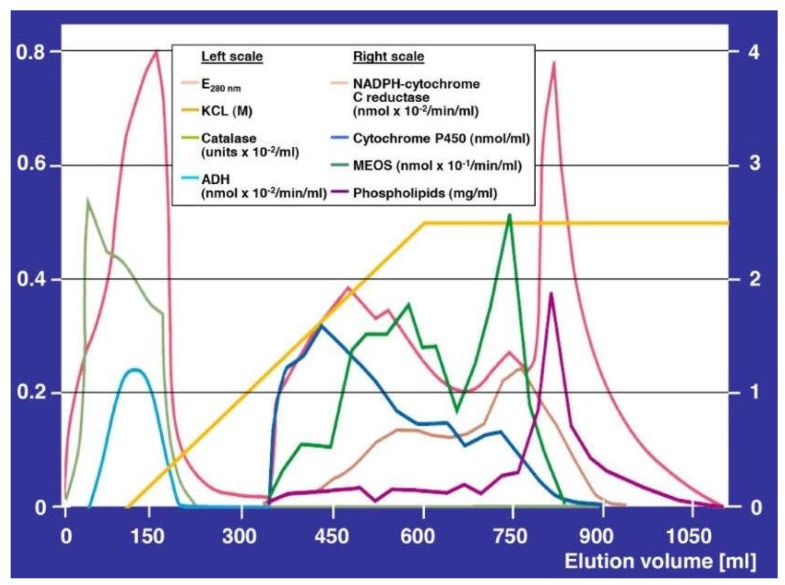
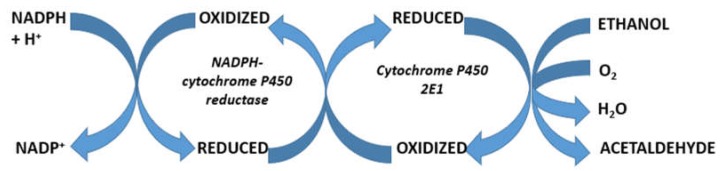
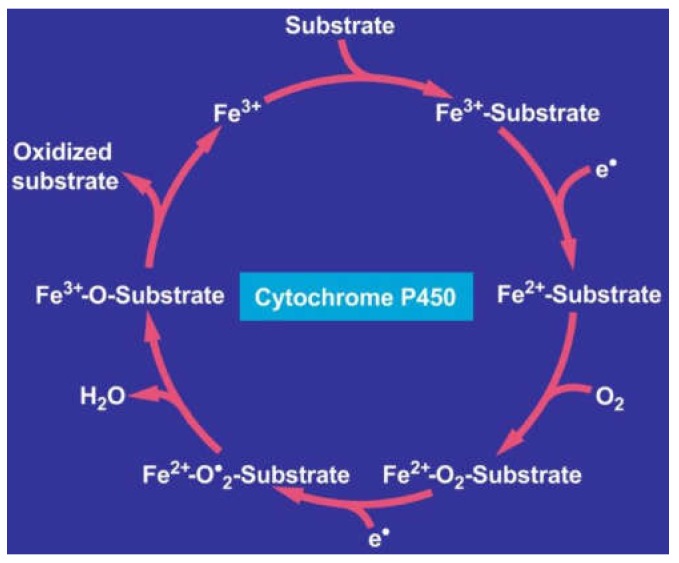
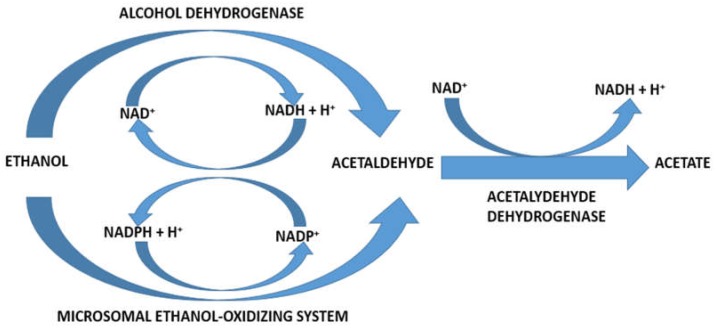
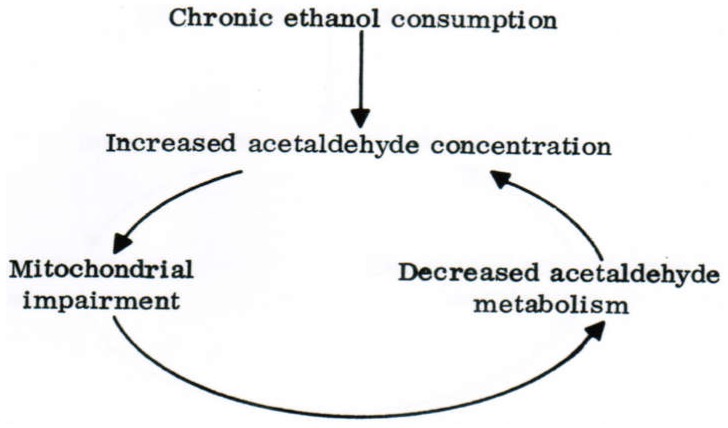
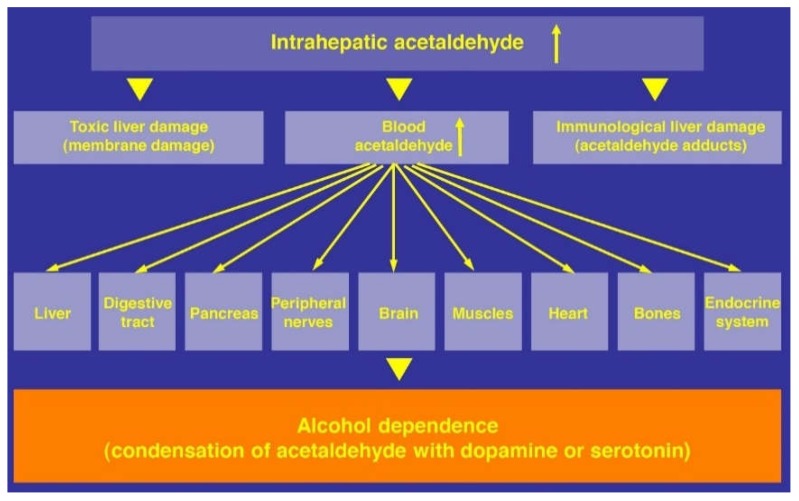
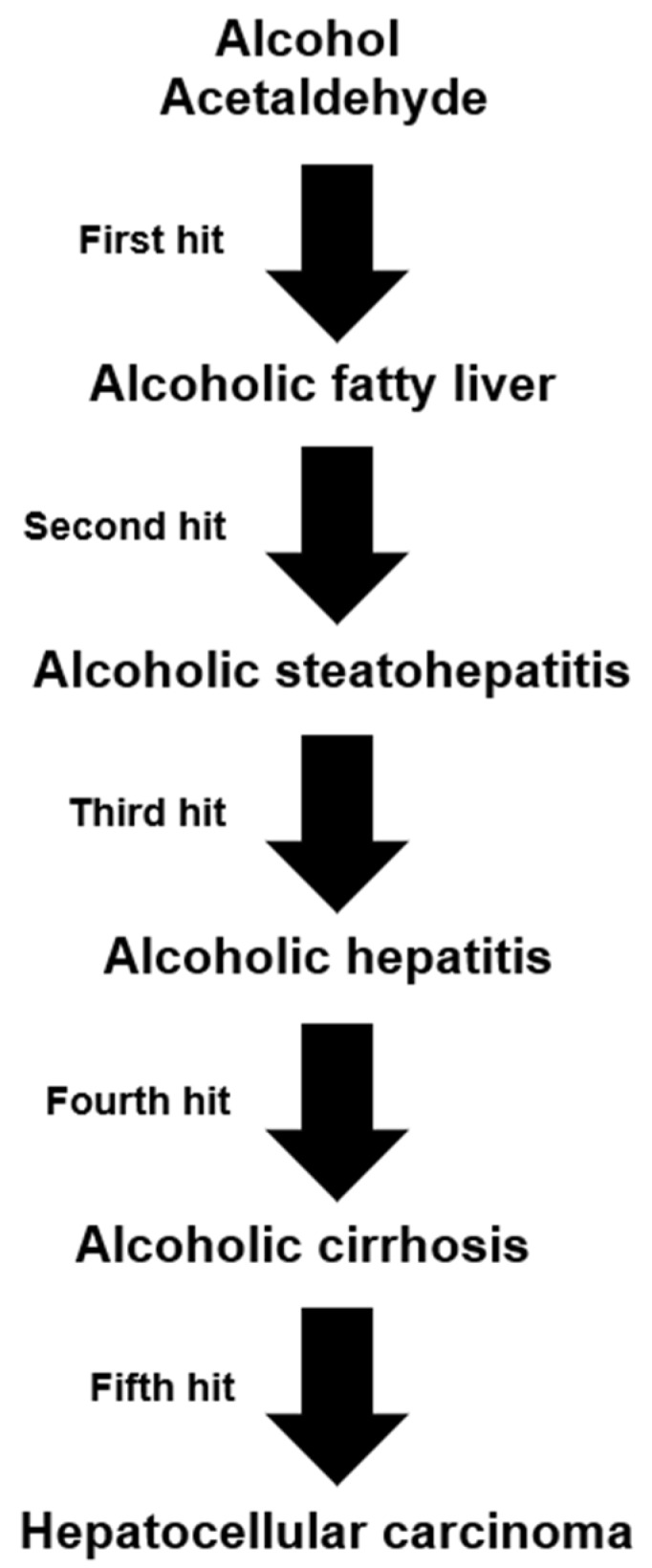
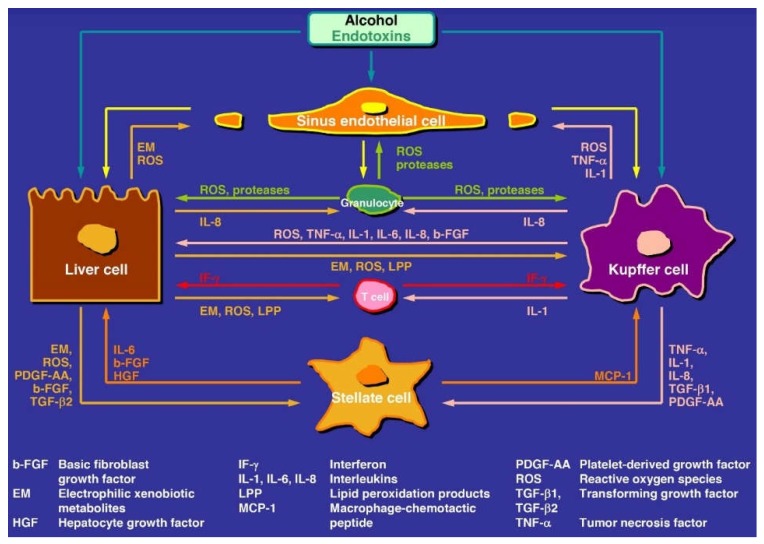
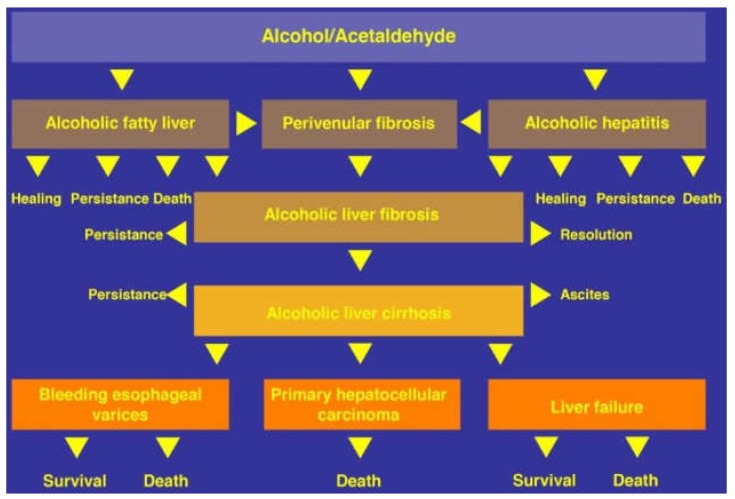
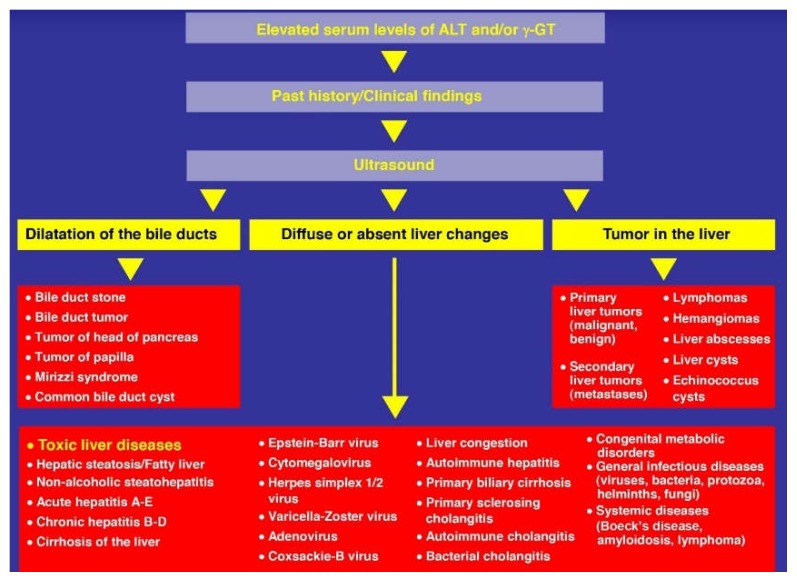
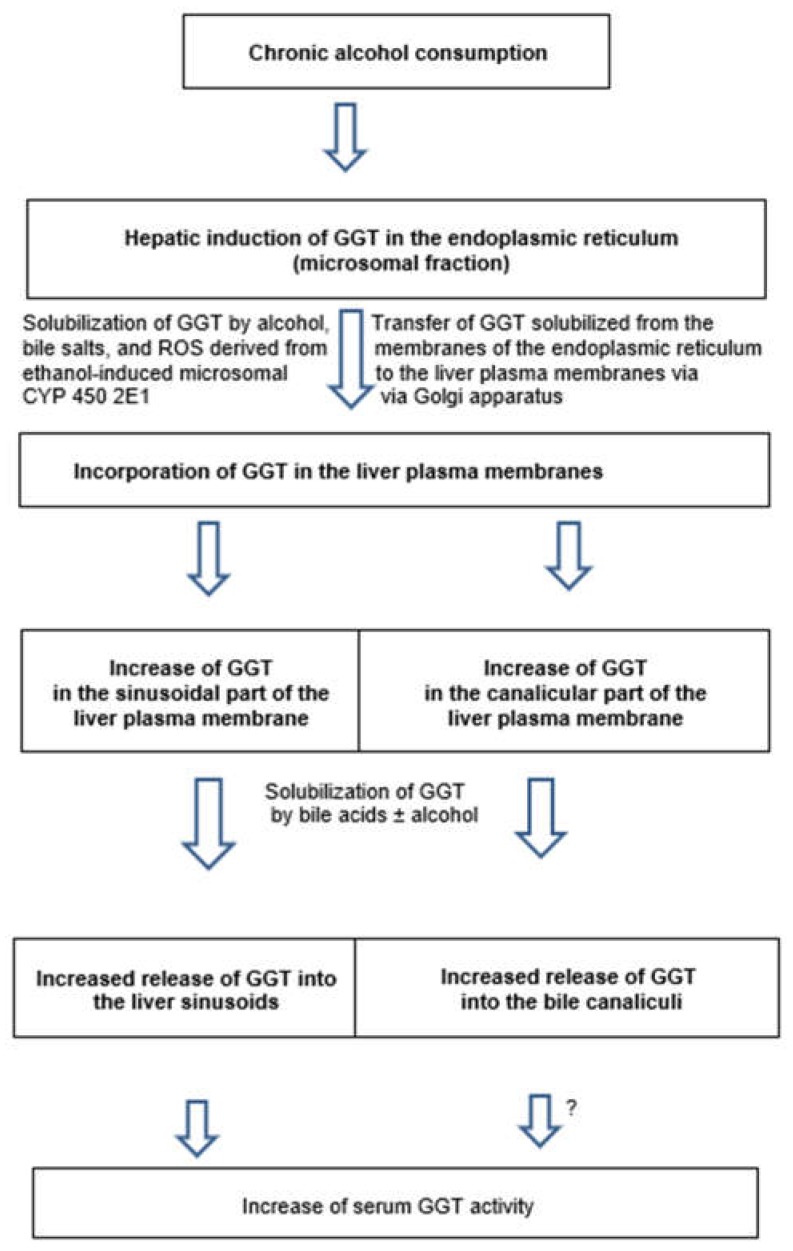
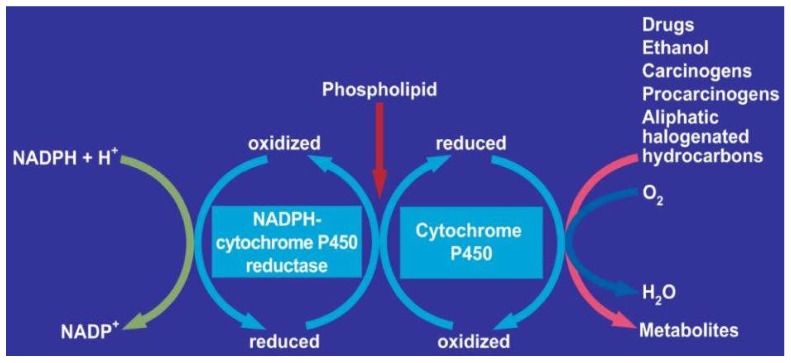
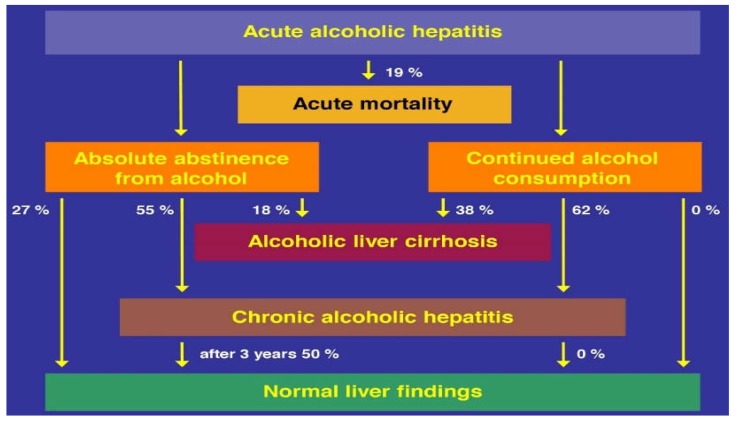
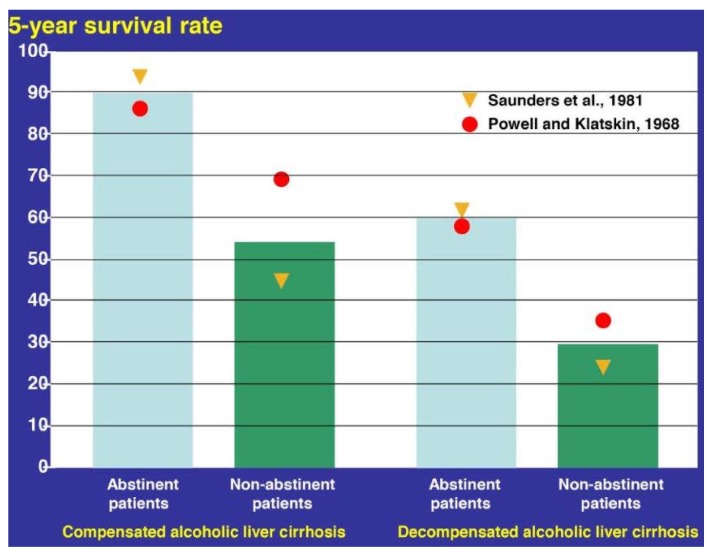
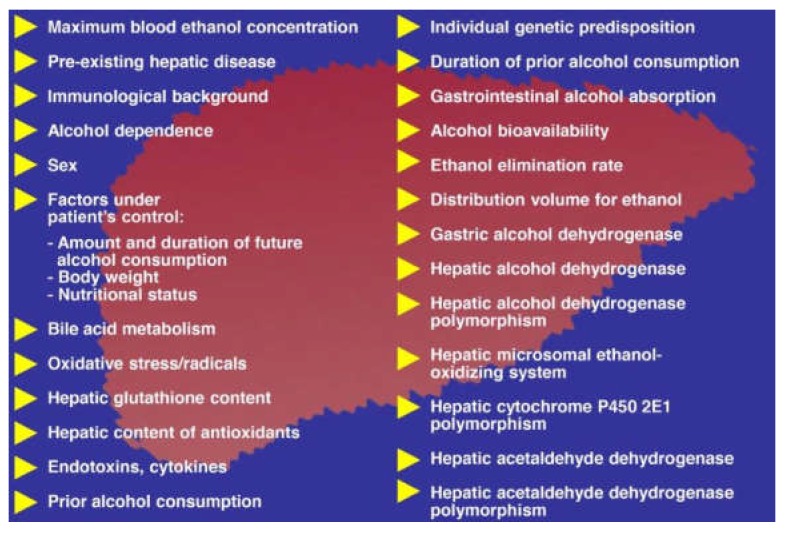
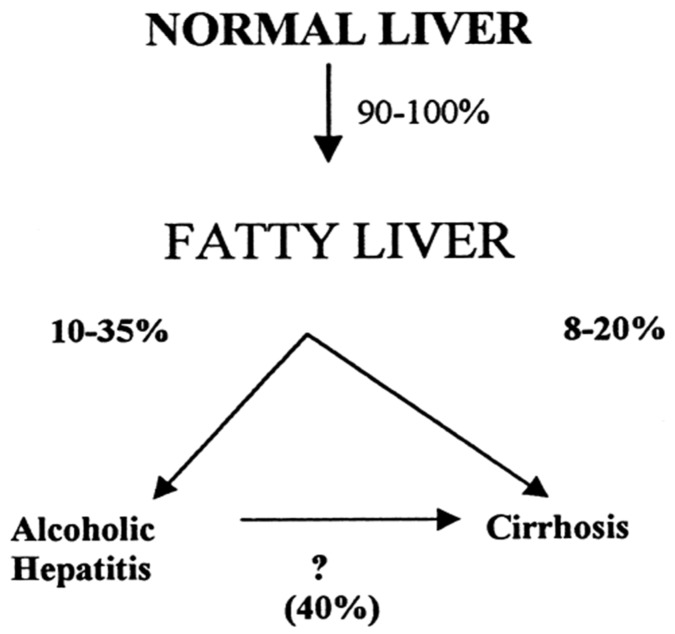
Alcoholic liver disease is the result of cascade events, which clinically first lead to alcoholic fatty liver, and then mostly via alcoholic steatohepatitis or alcoholic hepatitis potentially to cirrhosis and hepatocellular carcinoma. Pathogenetic events are linked to the metabolism of ethanol and acetaldehyde as its first oxidation product generated via hepatic alcohol dehydrogenase (ADH) and the microsomal ethanol-oxidizing system (MEOS), which depends on cytochrome P450 2E1 (CYP 2E1), and is inducible by chronic alcohol use. MEOS induction accelerates the metabolism of ethanol to acetaldehyde that facilitates organ injury including the liver, and it produces via CYP 2E1 many reactive oxygen species (ROS) such as ethoxy radical, hydroxyethyl radical, acetyl radical, singlet radical, superoxide radical, hydrogen peroxide, hydroxyl radical, alkoxyl radical, and peroxyl radical. These attack hepatocytes, Kupffer cells, stellate cells, and liver sinusoidal endothelial cells, and their signaling mediators such as interleukins, interferons, and growth factors, help to initiate liver injury including fibrosis and cirrhosis in susceptible individuals with specific risk factors. Through CYP 2E1-dependent ROS, more evidence is emerging that alcohol generates lipid peroxides and modifies the intestinal microbiome, thereby stimulating actions of endotoxins produced by intestinal bacteria; lipid peroxides and endotoxins are potential causes that are involved in alcoholic liver injury. Alcohol modifies SIRT1 (Sirtuin-1; derived from Silent mating type Information Regulation) and SIRT2, and most importantly, the innate and adapted immune systems, which may explain the individual differences of injury susceptibility. Metabolic pathways are also influenced by circadian rhythms, specific conditions known from living organisms including plants. Open for discussion is a 5-hit working hypothesis, attempting to define key elements involved in injury progression. In essence, although abundant biochemical mechanisms are proposed for the initiation and perpetuation of liver injury, patients with an alcohol problem benefit from permanent alcohol abstinence alone.
Keywords: CYP 2E1; MEOS; ROS; SIRT; acetaldehyde; alcohol dehydrogenase; alcohol metabolism; alcoholic cirrhosis; alcoholic fatty liver; alcoholic hepatitis; alcoholic liver disease; alcoholic steatohepatitis; circadian rhythms; endotoxins; ethanol; hepatocellular carcinoma; intestinal microbiome; microsomal ethanol-oxidizing system.
Conflict of interest statement
The author has no conflict of interest to declare, with respect to this invited review article.
Figures
References
-
- Sergent O., Djoudi-Aliche F., Lagadic-Gossmann D. Up-to date insight about membrane remodeling as a mechanism of action for ethanol-induced liver toxicity. In: Shimizu I., editor. Trends in Alcoholic Liver Disease–Clinical and Scientific Aspects. InTech; London, UK: [(accessed on 26 October 2018)]. Available online: https://cdn.intechopen.com/pdfs-wm/25884.pdf.
Publication types
LinkOut - more resources
Full Text Sources
