

Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury
- PMID: 30425237
- PMCID: PMC6233178
- DOI: 10.1038/s41419-018-1157-x
Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury
Abstract
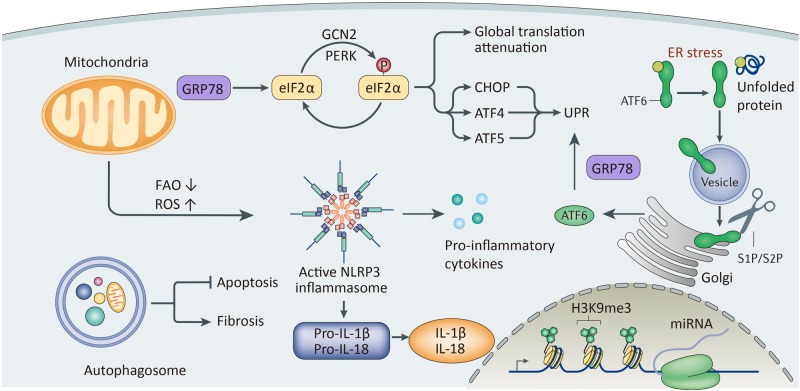
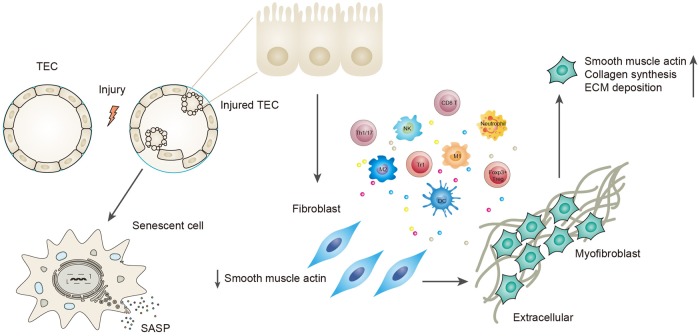
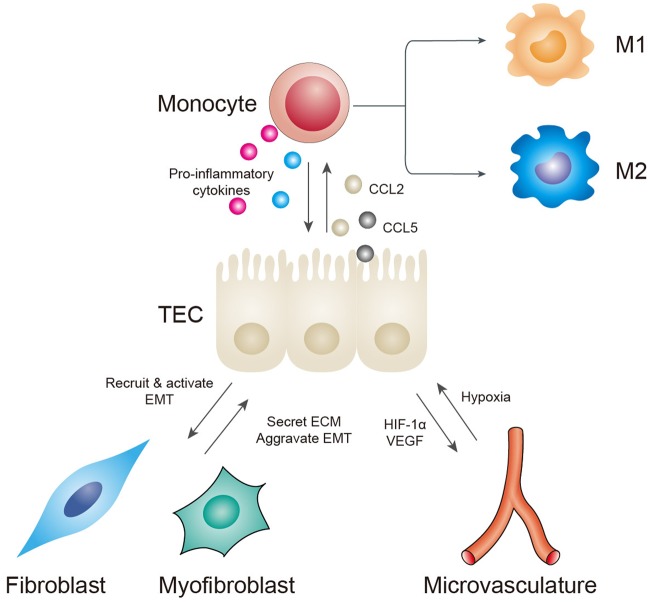
Renal fibrosis, especially tubulointerstitial fibrosis, is the inevitable outcome of all progressive chronic kidney diseases (CKDs) and exerts a great health burden worldwide. For a long time, interests in renal fibrosis have been concentrated on fibroblasts and myofibroblasts. However, in recent years, growing numbers of studies have focused on the role of tubular epithelial cells (TECs). TECs, rather than a victim or bystander, are probably a neglected mediator in renal fibrosis, responding to a variety of injuries. The maladaptive repair mechanisms of TECs may be the key point in this process. In this review, we will focus on the role of TECs in tubulointerstitial fibrosis. We will follow the fate of a tubular cell and depict the intracellular changes after injury. We will then discuss how the repair mechanism of tubular cells becomes maladaptive, and we will finally discuss the intercellular crosstalk in the interstitium that ultimately proceeds tubulointerstitial fibrosis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
