

TGFβ1 signaling sustains aryl hydrocarbon receptor (AHR) expression and restrains the pathogenic potential of TH17 cells by an AHR-independent mechanism
- PMID: 30425241
- PMCID: PMC6234206
- DOI: 10.1038/s41419-018-1107-7
TGFβ1 signaling sustains aryl hydrocarbon receptor (AHR) expression and restrains the pathogenic potential of TH17 cells by an AHR-independent mechanism
Abstract
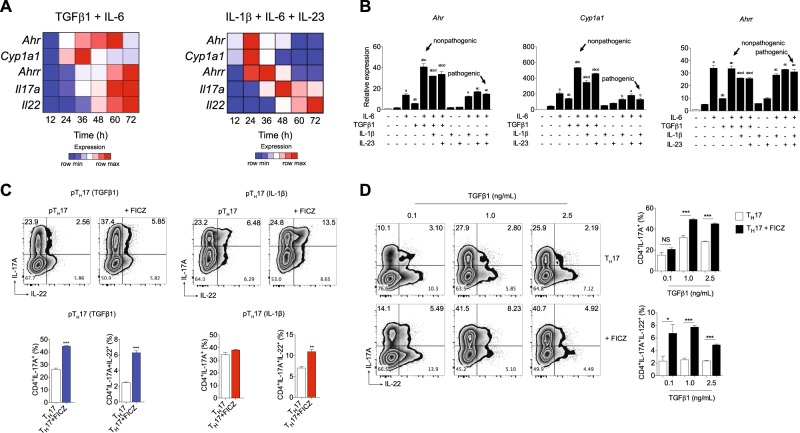
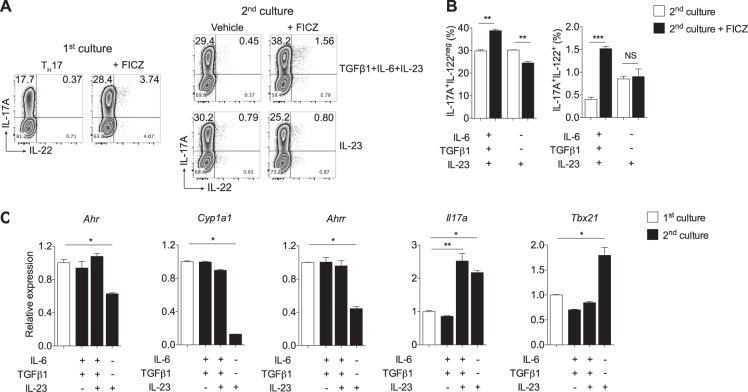
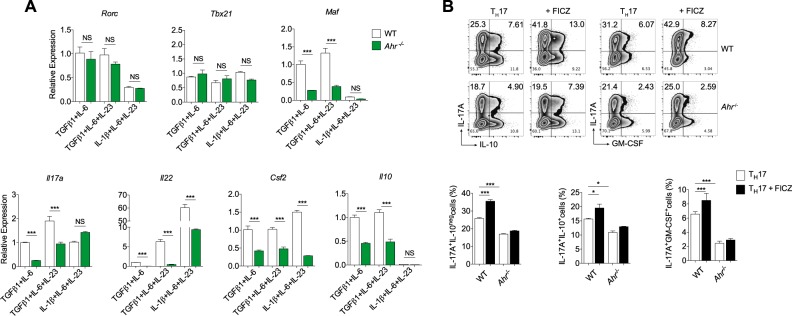
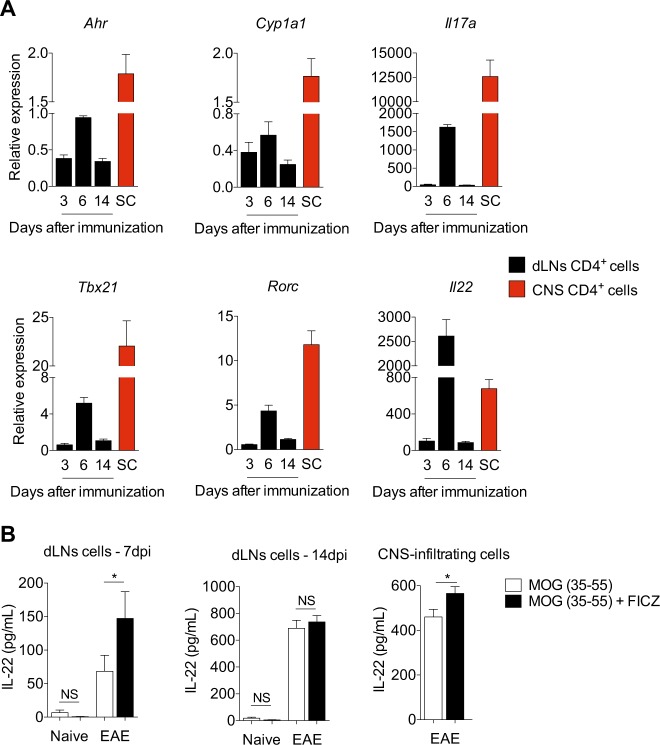
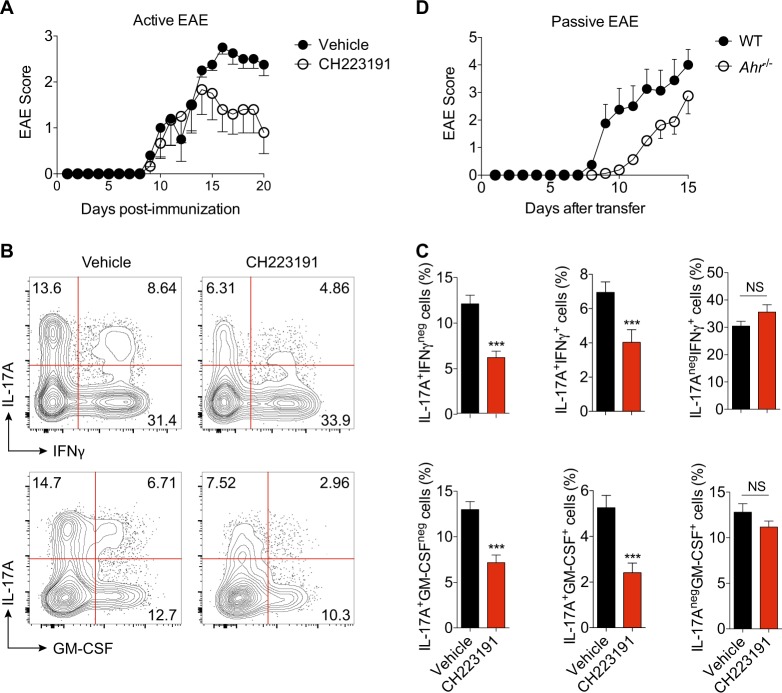
The aryl hydrocarbon receptor (AHR) is a transcription factor activated by ligand highly expressed on TH17 cells, and AHR-deficient CD4+ T cells have impaired production of IL-17A and IL-22. Although AHR activation can exacerbate in vivo TH17 cell-mediated autoimmunity, accumulating data indicate that AHR is a nonpathogenic TH17 marker. Thus it remains unclear how AHR activation is regulated and impacts on the generation of TH17 subsets. Here we demonstrated that AHR pathway is activated during in vitro pathogenic TH17 polarization, but it is quickly downregulated. Under these conditions, additional AHR activation promoted IL-22 but not IL-17A. Interestingly, AHR high sustained expression and IL-17A promotion were only achieved when TGFβ1 was present in the culture. In addition to the effect on AHR regulation, TGFβ1 presented a dual role by simultaneously suppressing the TH17 pathogenic phenotype acquisition. This latter effect was independent of AHR stimulation, since its activation did not confer a TH17 anti-inflammatory profile and Ahr-/- cells did not upregulate any TH17 pathogenic marker. Through the use of EAE model, we demonstrated that AHR is still functional in encephalitogenic CD4+ T cells and the adoptive transfer of Ahr-/- TH17 cells to recipient mice resulted in milder EAE development when compared to their WT counterparts. Altogether, our data demonstrated that although AHR is highly expressed on in vitro-generated nonpathogenic TH17 cells, its ligation does not shift TH17 cells to an anti-inflammatory phenotype. Further studies investigating the role of AHR beyond TH17 differentiation may provide a useful understanding of the physiopathology of autoimmune diseases.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
