

A web server for comparative analysis of single-cell RNA-seq data
- PMID: 30425249
- PMCID: PMC6233170
- DOI: 10.1038/s41467-018-07165-2
A web server for comparative analysis of single-cell RNA-seq data
Abstract
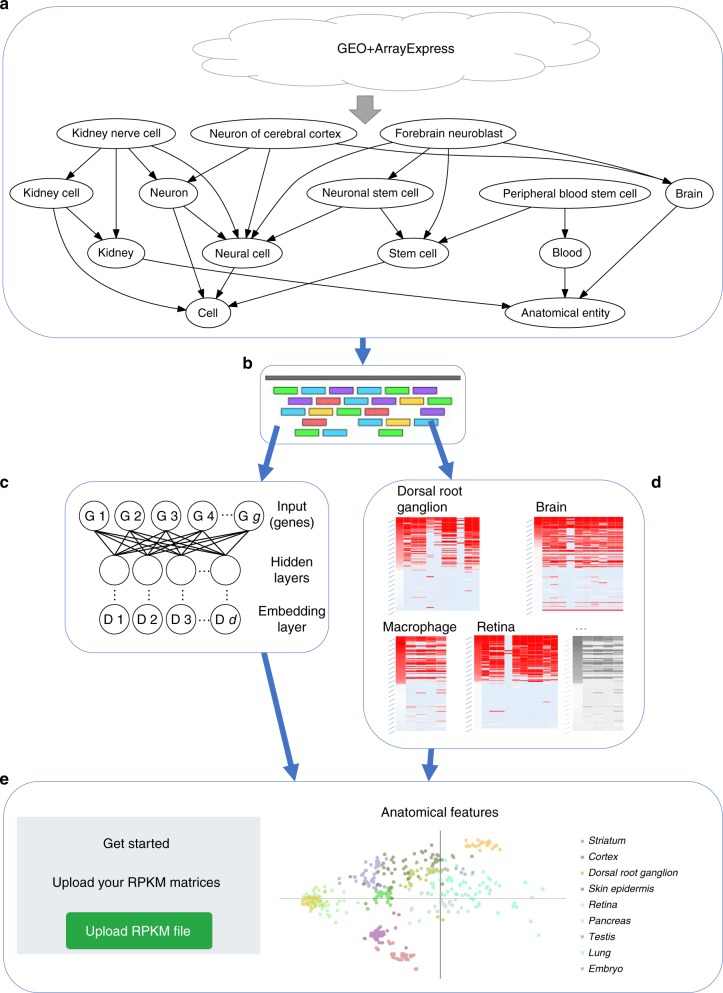
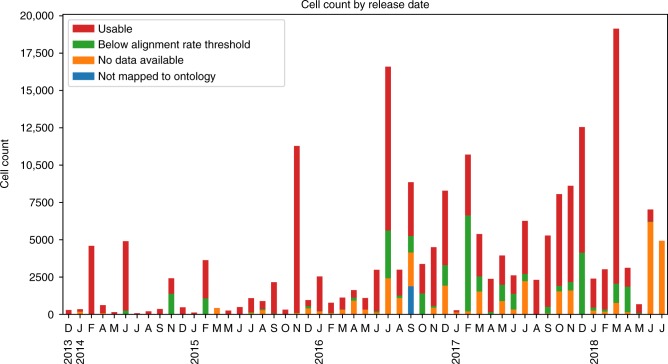
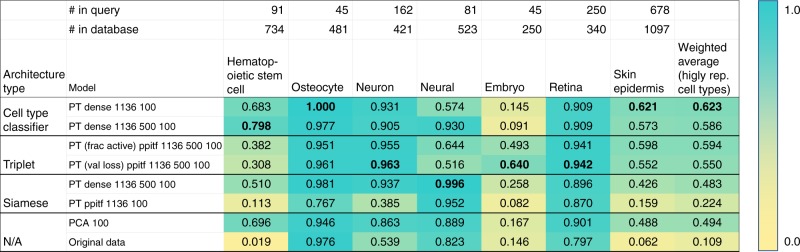
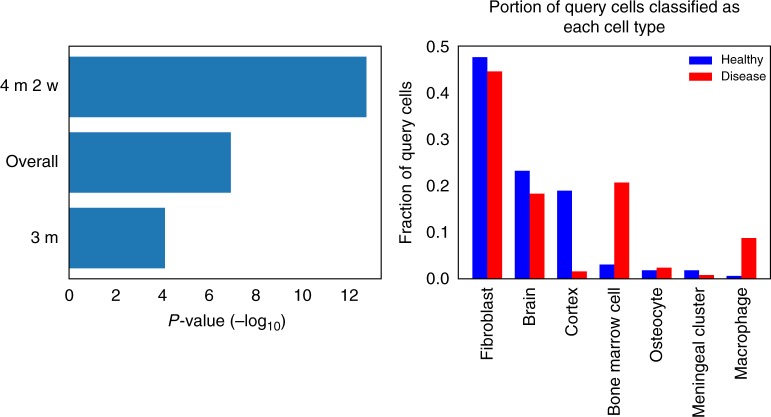
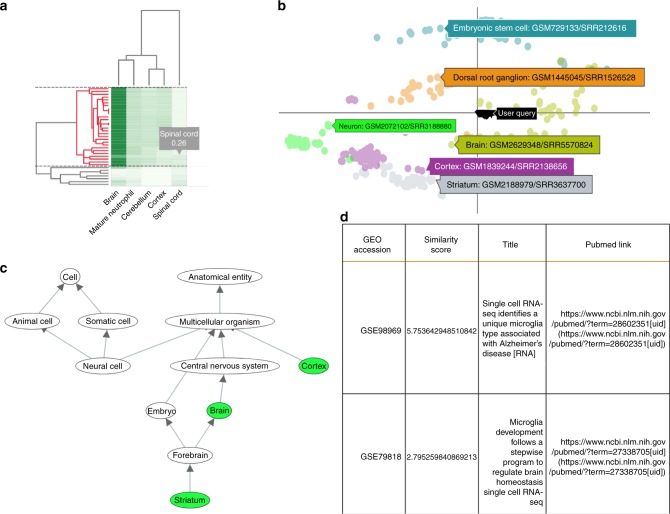
Single cell RNA-Seq (scRNA-seq) studies profile thousands of cells in heterogeneous environments. Current methods for characterizing cells perform unsupervised analysis followed by assignment using a small set of known marker genes. Such approaches are limited to a few, well characterized cell types. We developed an automated pipeline to download, process, and annotate publicly available scRNA-seq datasets to enable large scale supervised characterization. We extend supervised neural networks to obtain efficient and accurate representations for scRNA-seq data. We apply our pipeline to analyze data from over 500 different studies with over 300 unique cell types and show that supervised methods outperform unsupervised methods for cell type identification. A case study highlights the usefulness of these methods for comparing cell type distributions in healthy and diseased mice. Finally, we present scQuery, a web server which uses our neural networks and fast matching methods to determine cell types, key genes, and more.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Patel A. P., Tirosh I., Trombetta J. J., Shalek A. K., Gillespie S. M., Wakimoto H., Cahill D. P., Nahed B. V., Curry W. T., Martuza R. L., Louis D. N., Rozenblatt-Rosen O., Suva M. L., Regev A., Bernstein B. E. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. - DOI - PMC - PubMed
-
- Lescroart Fabienne, Wang Xiaonan, Lin Xionghui, Swedlund Benjamin, Gargouri Souhir, Sànchez-Dànes Adriana, Moignard Victoria, Dubois Christine, Paulissen Catherine, Kinston Sarah, Göttgens Berthold, Blanpain Cédric. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359(6380):1177–1181. doi: 10.1126/science.aao4174. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
