

Contribution of Host Genetics to the Variation of Microbial Composition of Cecum Lumen and Feces in Pigs
- PMID: 30429843
- PMCID: PMC6220110
- DOI: 10.3389/fmicb.2018.02626
Contribution of Host Genetics to the Variation of Microbial Composition of Cecum Lumen and Feces in Pigs
Abstract
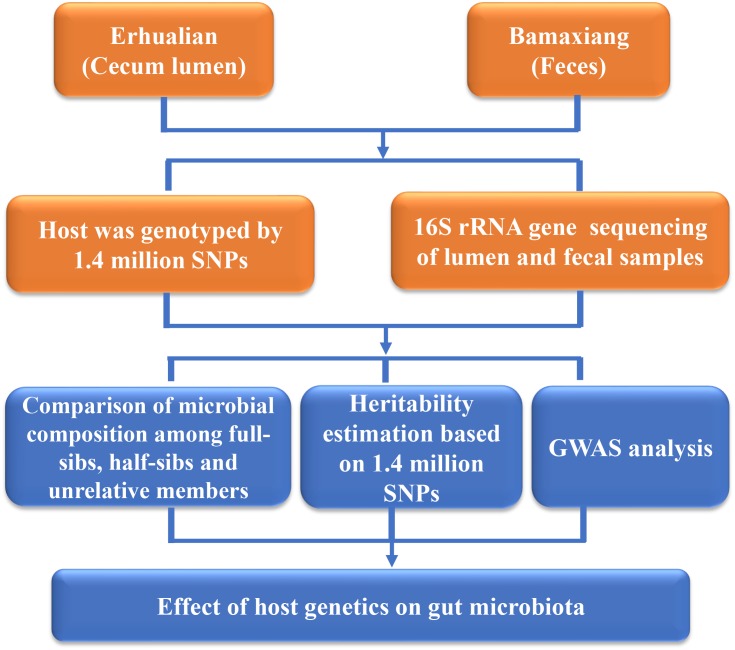
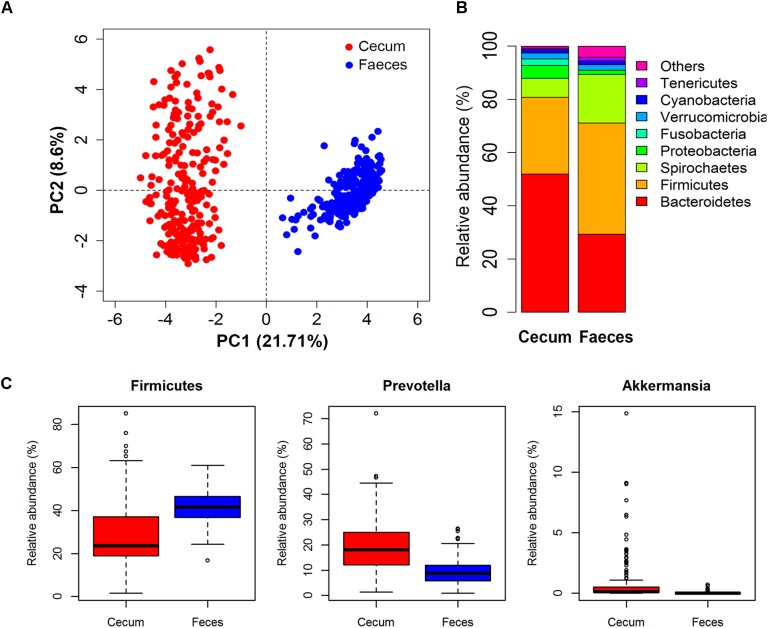
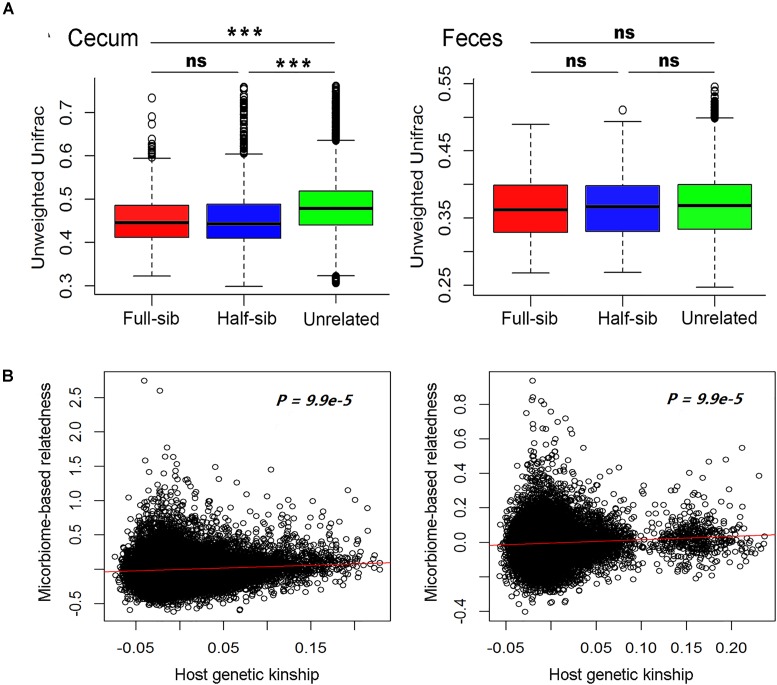
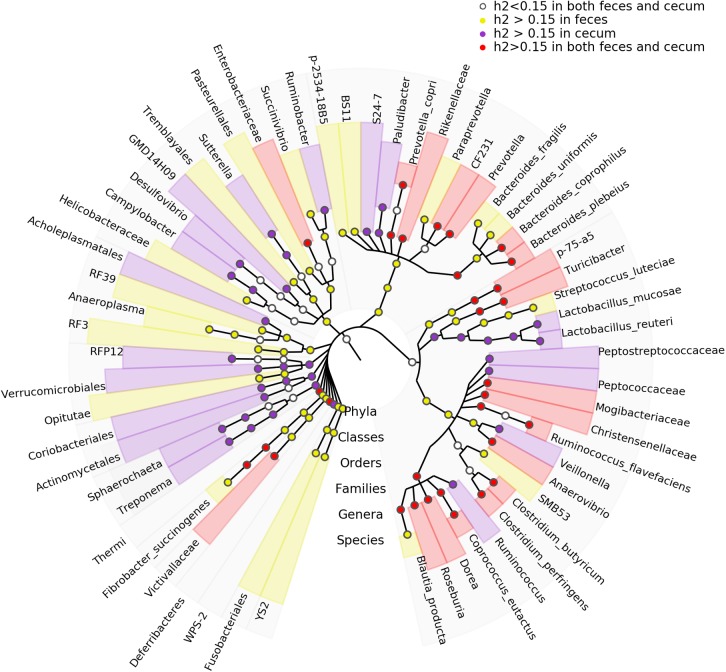
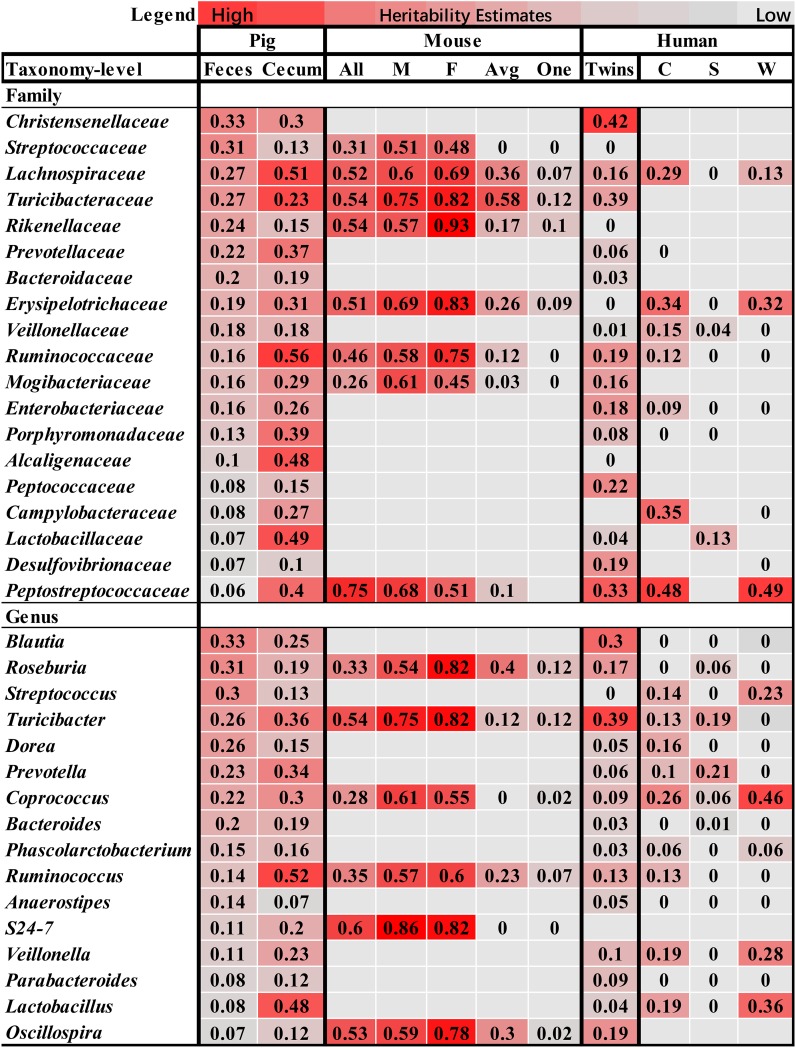
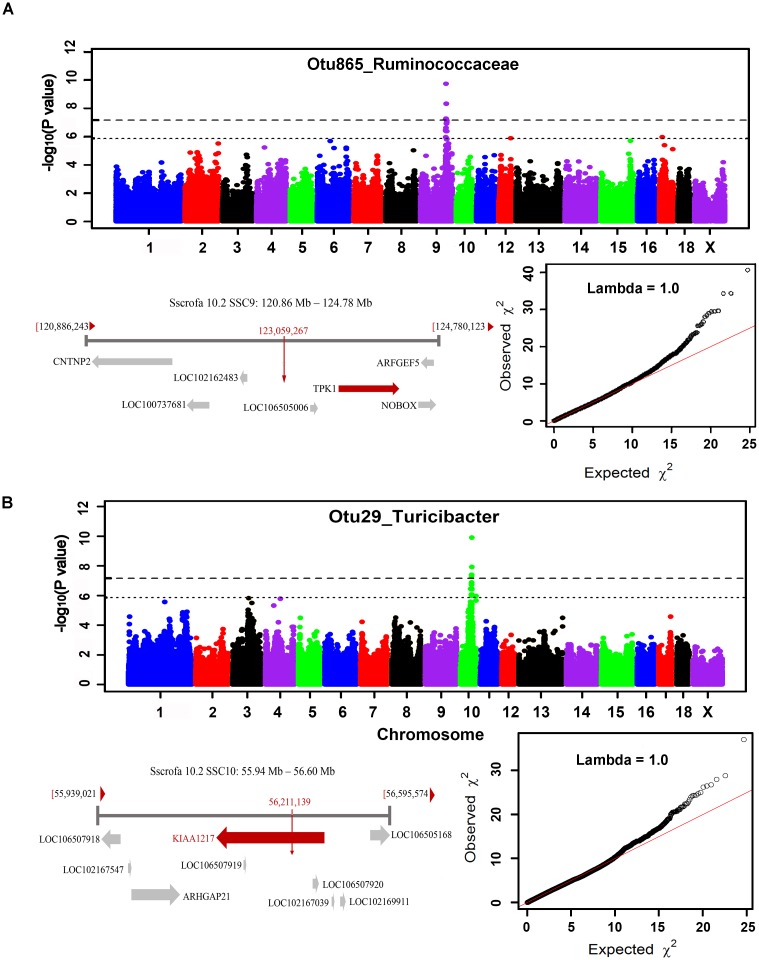
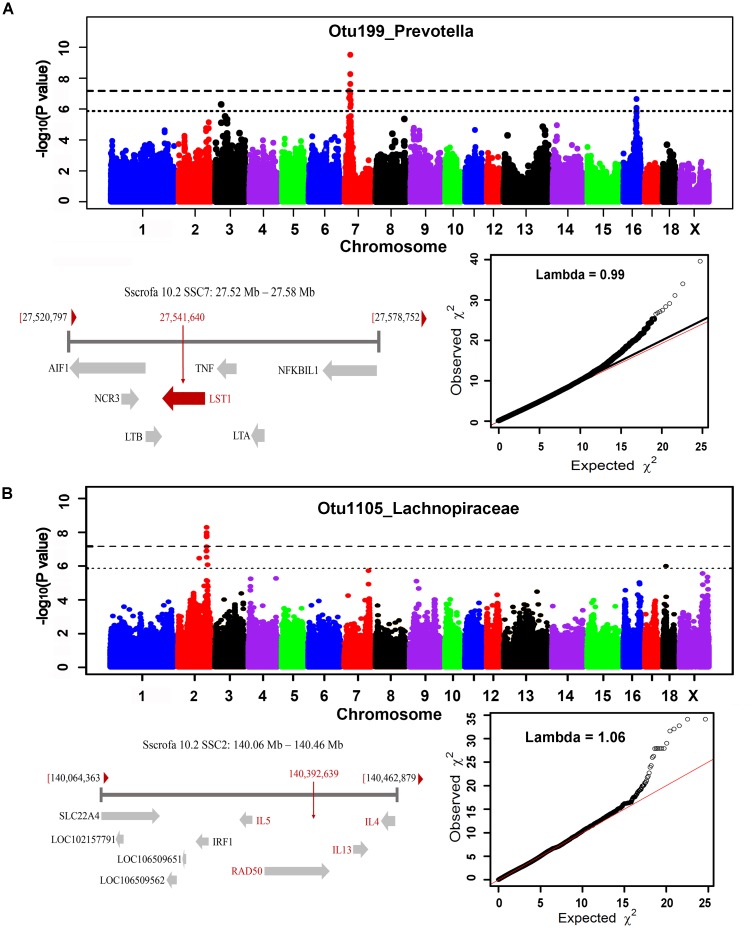
Pigs are a perfect model for studying the interaction between host genetics and gut microbiome due to the high similarity of gastrointestine and digestive system with humans, and the easily controlled feeding conditions. In this study, two pig populations which were raised in uniformed farm conditions and provided with the same commercial formula diet were used as the experimental animals. A systematical investigation of host genetic effect on the gut microbial composition was separately performed in porcine cecum lumen and feces samples through the comparison of microbial composition among full-sibs, half-sibs and unrelated members, heritability estimate (h 2), and genome-wide association study (GWAS). The results showed that full-sib members had a higher similarity of microbial composition than unrelated individuals. A significant correlation was observed between the microbial composition-based kinship and the host SNP-based kinship in both populations (P < 9.9 × 10-5). We identified 81 and 67 microbial taxa having h 2 > 0.15 in fecal and cecum luminal samples, respectively, including 31 taxa with h 2 > 0.15 in both types of samples. GWAS identified 40 and 34 significant associations between host genomic loci and the abundance or presence/absence of bacterial taxa in the fecal and cecum luminal samples. Functional classifications of host candidate genes related to microbial taxa are mainly associated with metabolism, immunity functions and response, and signal transduction. The high similarity of heritable taxa and functional categories of candidate genes among pig, human and mouse suggests the similar mechanism of the host genetic effect on gut microbiome across mammalian species. The results from this study provided another evidence that host genetics contributes significantly to the gut microbiome.
Keywords: candidate gene; genome-wide association study; gut microbiota; heritability estimate; host genetics; pigs.
Figures
References
-
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57 289–300.
-
- Benson A. K., Kelly S. A., Legge R., Ma F., Low S. J., Kim J., et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. U.S.A. 107 18933–18938. 10.1073/pnas.1007028107 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources