

Emerging Concepts and Therapies for Mucoobstructive Lung Disease
- PMID: 30431343
- PMCID: PMC6322026
- DOI: 10.1513/AnnalsATS.201806-368AW
Emerging Concepts and Therapies for Mucoobstructive Lung Disease
Abstract
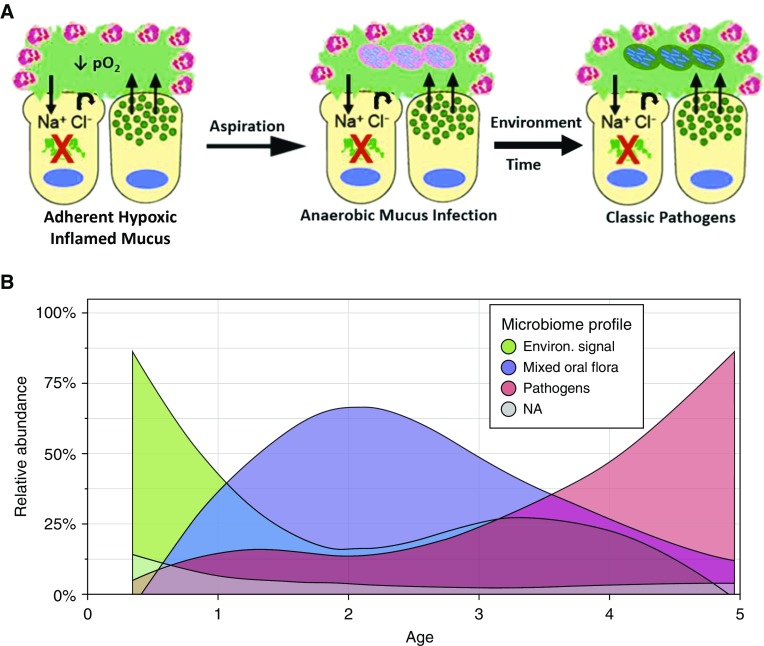
A spectrum of intrapulmonary airway diseases, for example, cigarette smoke-induced bronchitis, cystic fibrosis, primary ciliary dyskinesia, and non-cystic fibrosis bronchiectasis, can be categorized as "mucoobstructive" airway diseases. A common theme for these diseases appears to be the failure to properly regulate mucus concentration, producing mucus hyperconcentration that slows mucus transport and, importantly, generates plaque/plug adhesion to airway surfaces. These mucus plaques/plugs generate long diffusion distances for oxygen, producing hypoxic niches within adherent airway mucus and subjacent epithelia. Data suggest that concentrated mucus plaques/plugs are proinflammatory, in part mediated by release of IL-1α from hypoxic cells. The infectious component of mucoobstructive diseases may be initiated by anaerobic bacteria that proliferate within the nutrient-rich hypoxic mucus environment. Anaerobes ultimately may condition mucus to provide the environment for a succession to classic airway pathogens, including Staphylococcus aureus, Haemophilus influenzae, and ultimately Pseudomonas aeruginosa. Novel therapies to treat mucoobstructive diseases focus on restoring mucus concentration. Strategies to rehydrate mucus range from the inhalation of osmotically active solutes, designed to draw water into airway surfaces, to strategies designed to manipulate the relative rates of sodium absorption versus chloride secretion to endogenously restore epithelial hydration. Similarly, strategies designed to reduce the mucin burden in the airways, either by reducing mucin production/secretion or by clearing accumulated mucus (e.g., reducing agents), are under development. Thus, the new insights into a unifying process, that is, mucus hyperconcentration, that drives a significant component of the pathogenesis of mucoobstructive diseases promise multiple new therapeutic strategies to aid patients with this syndrome.
Keywords: IL-1α; anaerobes; hydration therapies; mucoobstruction; mucus hyperconcentration.
Figures


References
-
- Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
