

Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways
- PMID: 30431344
- PMCID: PMC6322034
- DOI: 10.1513/AnnalsATS.201802-144AW
Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways
Abstract
Idiopathic pulmonary fibrosis (IPF) is localized to the lung, is characterized by a pattern of heterogeneous, subpleural patches of fibrotic, remodeled lung, and is associated with a median survival of 3-5 years after diagnosis. A common gain-of-function MUC5B promoter variant, rs35705950, is the strongest risk factor (genetic and otherwise), accounting for at least 30% of the total risk of developing IPF. The MUC5B promoter variant can be used to identify individuals in the preclinical phase of this progressive disease, and, in the IPF lung, we have found that MUC5B is specifically overexpressed in bronchoalveolar epithelium. Thus, MUC5B represents a key molecule to understand the mechanisms that appear to initiate the fibroproliferative process in the bronchoalveolar epithelium. Moreover, focusing on MUC5B may provide a unique opportunity to define the early molecular events that lead to, and potentially prevent, the development of IPF.
Keywords: IPF; MUC5B; idiopathic pulmonary fibrosis.
Figures
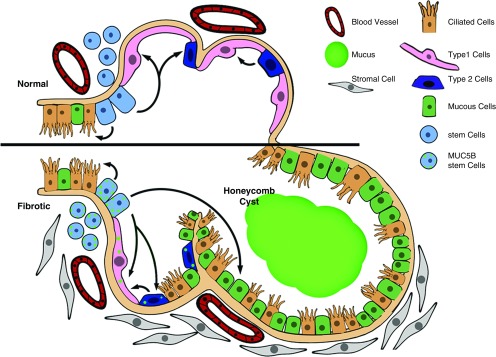
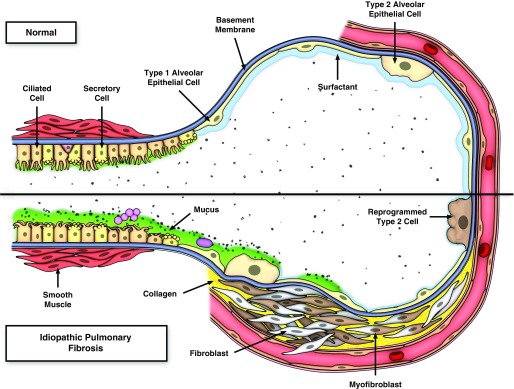
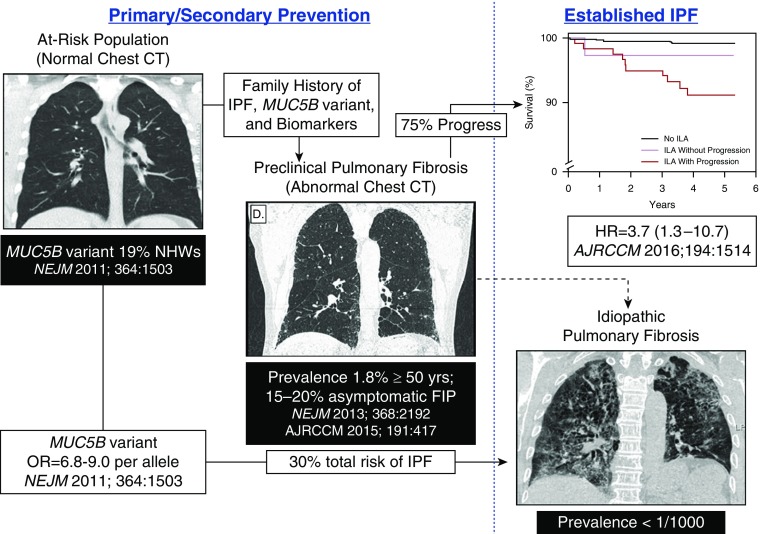
References
-
- American Thoracic Society International consensus statement idiopathic pulmonary fibrosis: diagnosis and treatment. Am J Respir Crit Care Med. 2000;161:646–664. - PubMed
-
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–248. - PubMed
-
- Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–284. - PubMed
-
- Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thorac Soc. 2014;11:1176–1185. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
