

Mucociliary Transport in Healthy and Cystic Fibrosis Pig Airways
- PMID: 30431346
- PMCID: PMC6322029
- DOI: 10.1513/AnnalsATS.201805-308AW
Mucociliary Transport in Healthy and Cystic Fibrosis Pig Airways
Abstract
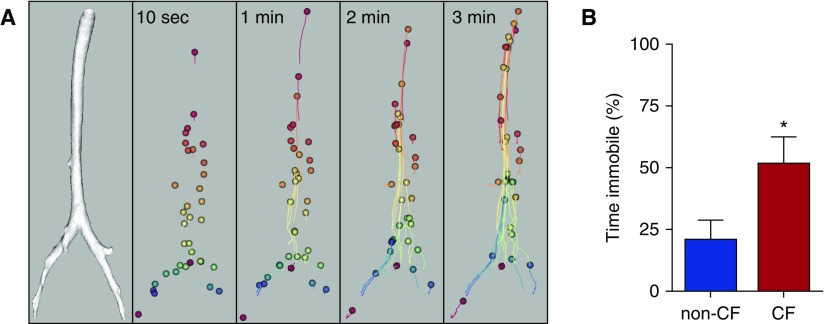
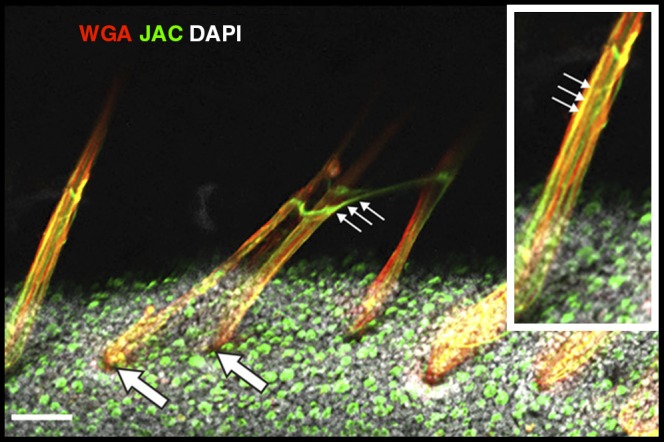
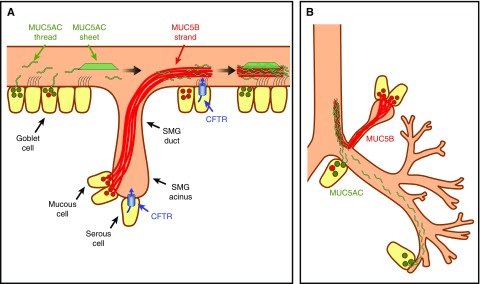
Cystic fibrosis (CF) lung disease is the major cause of morbidity and mortality in people with CF. Abnormal mucociliary transport has been the leading hypothesis for the underlying pathogenesis of CF airway disease. However, this has been difficult to investigate at very early time points. A porcine CF model, which recapitulates many features of CF disease in humans, enables studies to be performed in non-CF and CF pigs on the day that they are born. In newborn CF pigs, we found that under basal conditions, mucociliary transport rates in non-CF and CF pigs are similar. However, after cholinergic stimulation, which stimulates submucosal gland secretion, particles become stuck in the CF airways owing to a failure of mucus strands to release from submucosal glands. In this review, we summarize these recent discoveries and also discuss the morphology, composition, and function of mucins in the porcine lung.
Keywords: cystic fibrosis; mucociliary transport; mucus; submucosal gland.
Figures
Similar articles
-
Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis.Science. 2014 Aug 15;345(6198):818-22. doi: 10.1126/science.1255825. Science. 2014. PMID: 25124441 Free PMC article.
-
Acid exposure disrupts mucus secretion and impairs mucociliary transport in neonatal piglet airways.Am J Physiol Lung Cell Mol Physiol. 2020 May 1;318(5):L873-L887. doi: 10.1152/ajplung.00025.2020. Epub 2020 Mar 11. Am J Physiol Lung Cell Mol Physiol. 2020. PMID: 32160007 Free PMC article.
-
Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs.Proc Natl Acad Sci U S A. 2022 Jan 25;119(4):e2119759119. doi: 10.1073/pnas.2119759119. Proc Natl Acad Sci U S A. 2022. PMID: 35046051 Free PMC article.
-
HCO3- transport in relation to mucus secretion from submucosal glands.JOP. 2001 Jul;2(4 Suppl):280-4. JOP. 2001. PMID: 11875272 Review.
-
Airway surface dehydration in cystic fibrosis: pathogenesis and therapy.Annu Rev Med. 2007;58:157-70. doi: 10.1146/annurev.med.58.071905.105316. Annu Rev Med. 2007. PMID: 17217330 Review.
Cited by
-
Dynamic regulation of airway surface liquid pH by TMEM16A and SLC26A4 in cystic fibrosis nasal epithelia with rare mutations.Proc Natl Acad Sci U S A. 2023 Nov 21;120(47):e2307551120. doi: 10.1073/pnas.2307551120. Epub 2023 Nov 15. Proc Natl Acad Sci U S A. 2023. PMID: 37967223 Free PMC article.
-
Normal murine respiratory tract has its mucus concentrated in clouds based on the Muc5b mucin.Am J Physiol Lung Cell Mol Physiol. 2020 Jun 1;318(6):L1270-L1279. doi: 10.1152/ajplung.00485.2019. Epub 2020 Apr 29. Am J Physiol Lung Cell Mol Physiol. 2020. PMID: 32348677 Free PMC article.
-
Factoring in the Complexity of the Cystic Fibrosis Lung to Understand Aspergillus fumigatus and Pseudomonas aeruginosa Interactions.Pathogens. 2020 Aug 6;9(8):639. doi: 10.3390/pathogens9080639. Pathogens. 2020. PMID: 32781694 Free PMC article. Review.
-
The pH-sensitive translocation of V-ATPase in the small airway of cystic fibrosis pigs.Am J Physiol Lung Cell Mol Physiol. 2024 Nov 1;327(5):L749-L755. doi: 10.1152/ajplung.00147.2024. Epub 2024 Sep 10. Am J Physiol Lung Cell Mol Physiol. 2024. PMID: 39254087 Free PMC article.
-
An ex vivo rat trachea model reveals abnormal airway physiology and a gland secretion defect in cystic fibrosis.PLoS One. 2023 Oct 24;18(10):e0293367. doi: 10.1371/journal.pone.0293367. eCollection 2023. PLoS One. 2023. PMID: 37874846 Free PMC article.
References
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR. et al. Cystic fibrosis. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, editors; The metabolic and molecular basis of inherited disease, 8th ed. New York: McGraw-Hill; 2001. pp. 5121–5189.
-
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. - PubMed
-
- Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol. 1995;20:63–70. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
