

Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics
- PMID: 30441796
- PMCID: PMC6274730
- DOI: 10.3390/ijms19113594
Risk Factors and Pathogenesis of HIV-Associated Neurocognitive Disorder: The Role of Host Genetics
Abstract
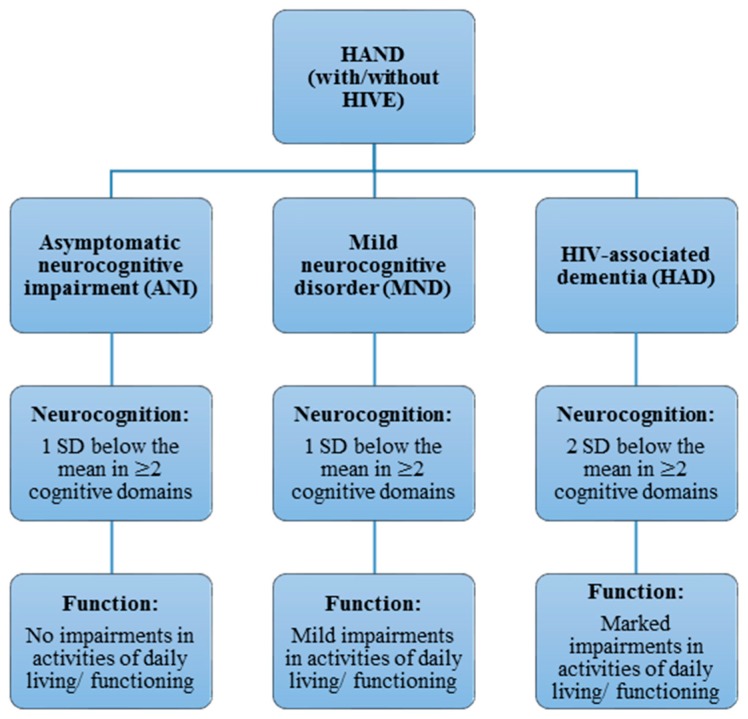
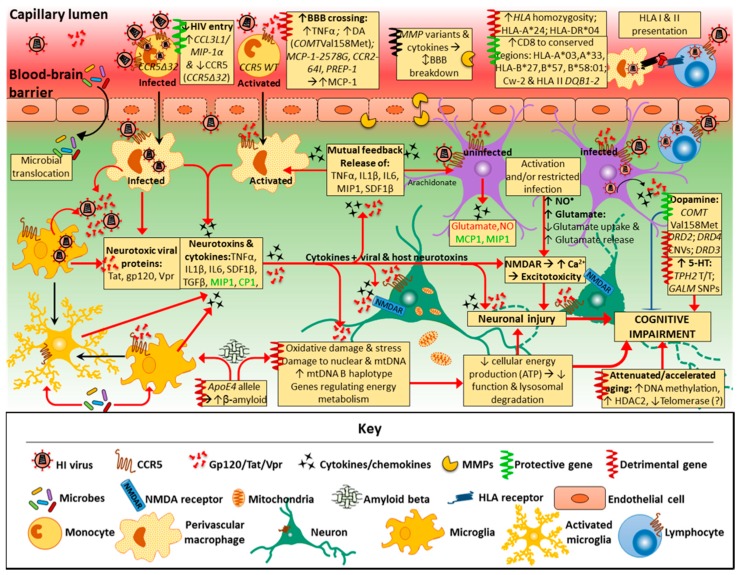
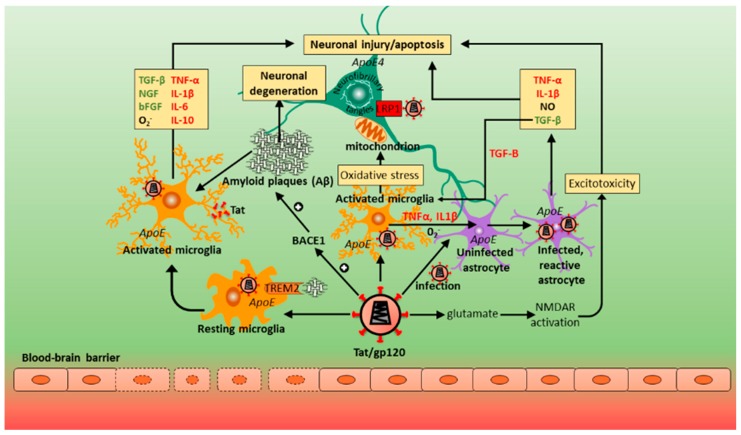
Neurocognitive impairments associated with human immunodeficiency virus (HIV) infection remain a considerable health issue for almost half the people living with HIV, despite progress in HIV treatment through combination antiretroviral therapy (cART). The pathogenesis and risk factors of HIV-associated neurocognitive disorder (HAND) are still incompletely understood. This is partly due to the complexity of HAND diagnostics, as phenotypes present with high variability and change over time. Our current understanding is that HIV enters the central nervous system (CNS) during infection, persisting and replicating in resident immune and supporting cells, with the subsequent host immune response and inflammation likely adding to the development of HAND. Differences in host (human) genetics determine, in part, the effectiveness of the immune response and other factors that increase the vulnerability to HAND. This review describes findings from studies investigating the role of human host genetics in the pathogenesis of HAND, including potential risk factors for developing HAND. The similarities and differences between HAND and Alzheimer's disease are also discussed. While some specific variations in host genes regulating immune responses and neurotransmission have been associated with protection or risk of HAND development, the effects are generally small and findings poorly replicated. Nevertheless, a few specific gene variants appear to affect the risk for developing HAND and aid our understanding of HAND pathogenesis.
Keywords: HIV; HIV-associated dementia; HIV-associated neurocognitive disorders; HIV-encephalitis; cognitive impairment; host genetics; neuroAIDS; polymorphisms; risk factors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
The role of host genetics in the susceptibility for HIV-associated neurocognitive disorders.AIDS Behav. 2009 Feb;13(1):118-32. doi: 10.1007/s10461-008-9360-x. Epub 2008 Feb 9. AIDS Behav. 2009. PMID: 18264751 Free PMC article. Review.
-
Host genetic factors predisposing to HIV-associated neurocognitive disorder.Curr HIV/AIDS Rep. 2014 Sep;11(3):336-52. doi: 10.1007/s11904-014-0222-z. Curr HIV/AIDS Rep. 2014. PMID: 24996618 Free PMC article. Review.
-
Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications.Brain Behav Immun. 2015 Mar;45:1-12. doi: 10.1016/j.bbi.2014.10.008. Epub 2014 Oct 22. Brain Behav Immun. 2015. PMID: 25449672 Free PMC article. Review.
-
PET brain imaging in HIV-associated neurocognitive disorders (HAND) in the era of combination antiretroviral therapy.Eur J Nucl Med Mol Imaging. 2017 May;44(5):895-902. doi: 10.1007/s00259-016-3602-3. Epub 2017 Jan 5. Eur J Nucl Med Mol Imaging. 2017. PMID: 28058461 Review.
-
Biotypes of HIV-associated neurocognitive disorders based on viral and immune pathogenesis.Curr Opin Infect Dis. 2022 Jun 1;35(3):223-230. doi: 10.1097/QCO.0000000000000825. Curr Opin Infect Dis. 2022. PMID: 35665716 Free PMC article. Review.
Cited by
-
7,8-Dihydroxyflavone improves neuropathological changes in the brain of Tg26 mice, a model for HIV-associated neurocognitive disorder.Sci Rep. 2021 Sep 16;11(1):18519. doi: 10.1038/s41598-021-97220-8. Sci Rep. 2021. PMID: 34531413 Free PMC article.
-
Astrocytes, HIV and the Glymphatic System: A Disease of Disrupted Waste Management?Front Cell Infect Microbiol. 2020 Sep 29;10:523379. doi: 10.3389/fcimb.2020.523379. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 33134185 Free PMC article. Review.
-
The Impact of Cannabis Use on Cognition in People with HIV: Evidence of Function-Dependent Effects and Mechanisms from Clinical and Preclinical Studies.Curr HIV/AIDS Rep. 2024 Jun;21(3):87-115. doi: 10.1007/s11904-024-00698-w. Epub 2024 Apr 11. Curr HIV/AIDS Rep. 2024. PMID: 38602558 Free PMC article.
-
HIV infection in microglia leads to senescence, triggering activation of neurotoxicity pathways.bioRxiv [Preprint]. 2025 May 8:2025.05.08.651477. doi: 10.1101/2025.05.08.651477. bioRxiv. 2025. PMID: 40654822 Free PMC article. Preprint.
-
Efficacy of aerobic exercise for HIV-associated neurocognitive disorders receiving ART: An RCT.S Afr J Physiother. 2024 Dec 11;80(1):2104. doi: 10.4102/sajp.v80i1.2104. eCollection 2024. S Afr J Physiother. 2024. PMID: 39822344 Free PMC article.
References
-
- Danforth K., Granich R., Wiedeman D., Baxi S., Padian N. Global mortality and morbidity of HIV/AIDS. In: Holmes K.K., Bertozzi S., Bloom B.R., Jha P., editors. Major Infectious Diseases. World Health Organization; Washington, DC, USA: 2017. - PubMed
-
- Arendt G., Grauer O., Hahn K., Maschke M., Obermann M., Husstedt I. Neues bei HIV und neuro-AIDS. Aktuelle Neurol. 2015;42:445–455. doi: 10.1055/s-0035-1552692. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
