

Mollusc genomes reveal variability in patterns of LTR-retrotransposons dynamics
- PMID: 30442098
- PMCID: PMC6238403
- DOI: 10.1186/s12864-018-5200-1
Mollusc genomes reveal variability in patterns of LTR-retrotransposons dynamics
Abstract
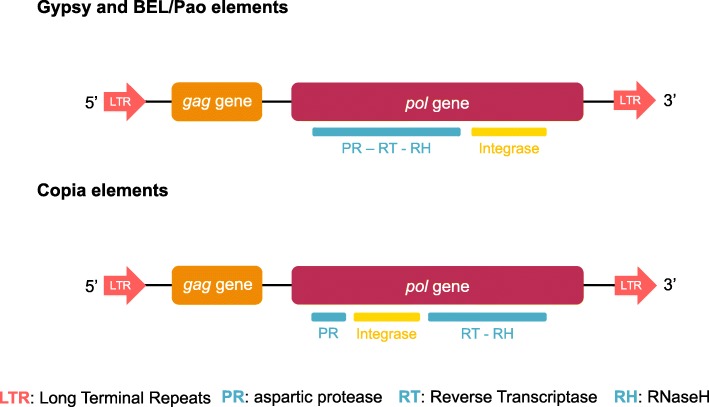
Background: The three superfamilies of Long Terminal Repeat (LTR) retrotransposons are a widespread kind of transposable element and a major factor in eukaryotic genome evolution. In metazoans, recent studies suggested that Copia LTR-retrotransposons display specific dynamic compared to the more abundant and diverse Gypsy elements. Indeed, Copia elements show a relative scarcity and the prevalence of only a few clades in specific hosts. Thus, BEL/Pao seems to be the second most abundant superfamily. However, the generality of these assumptions remains to be assessed. Therefore, we carried out the first large-scale comparative genomic analysis of LTR-retrotransposons in molluscs. The aim of this study was to analyse the diversity, copy numbers, genomic proportions and distribution of LTR-retrotransposons in a large host phylum.
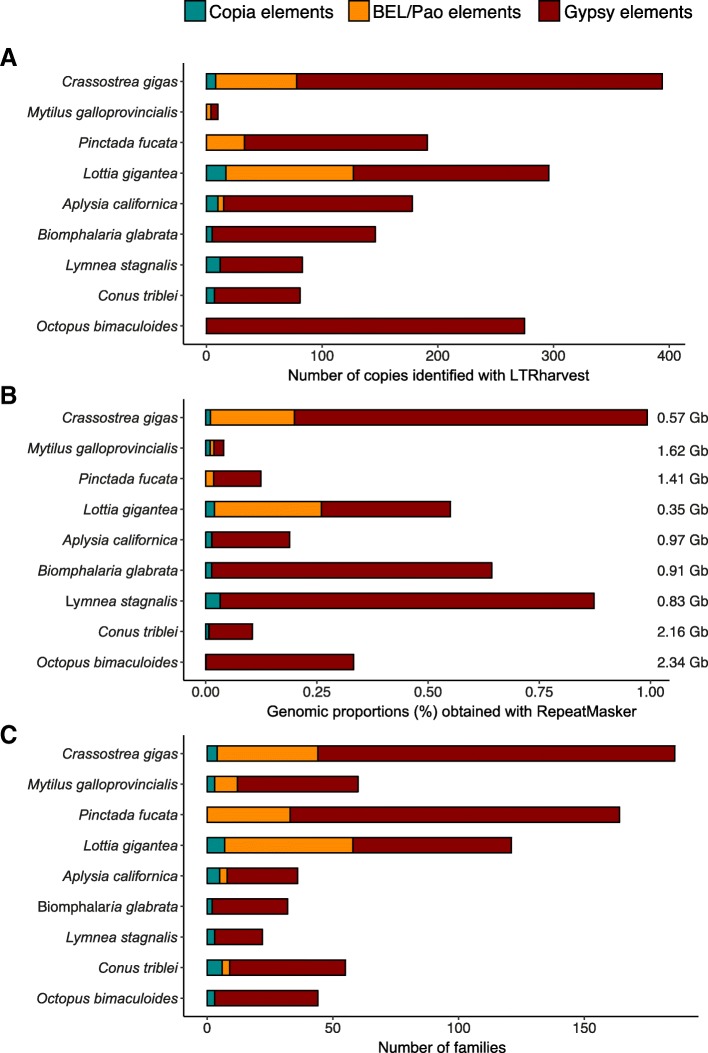
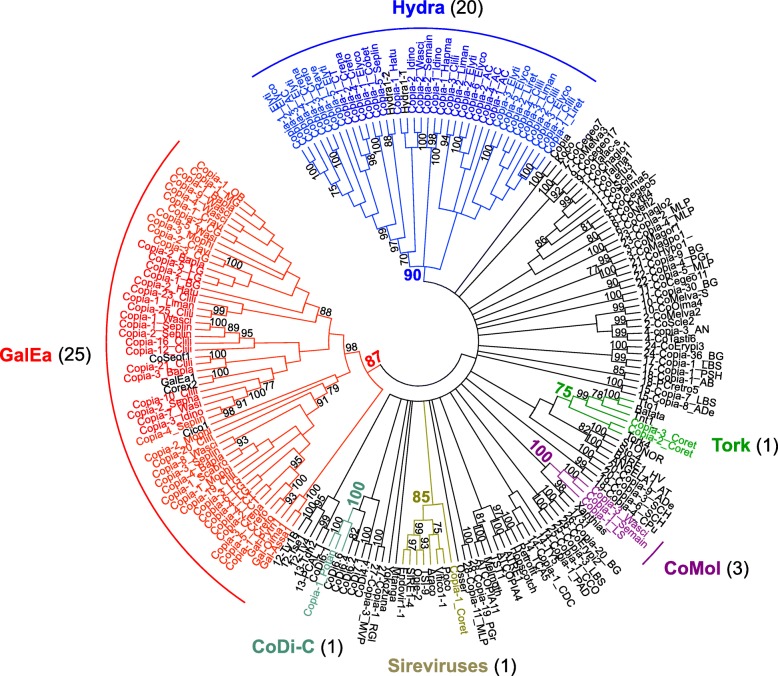
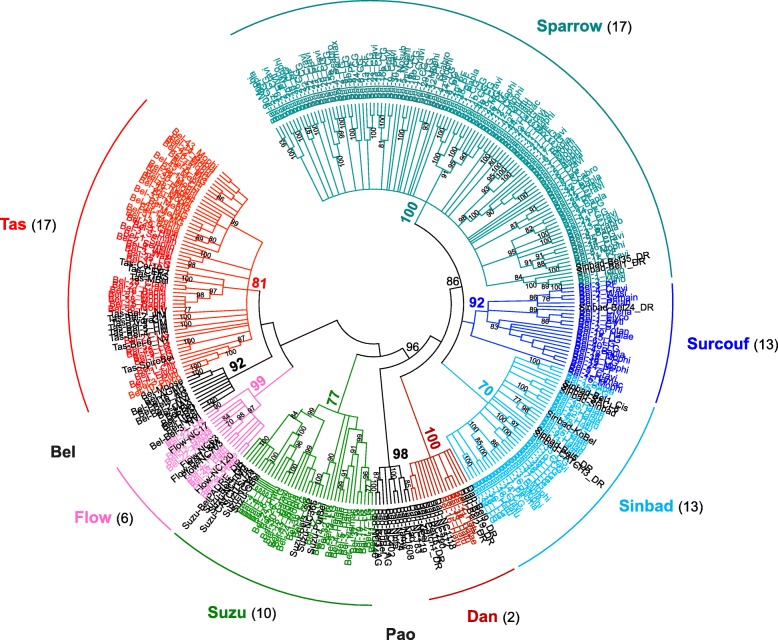
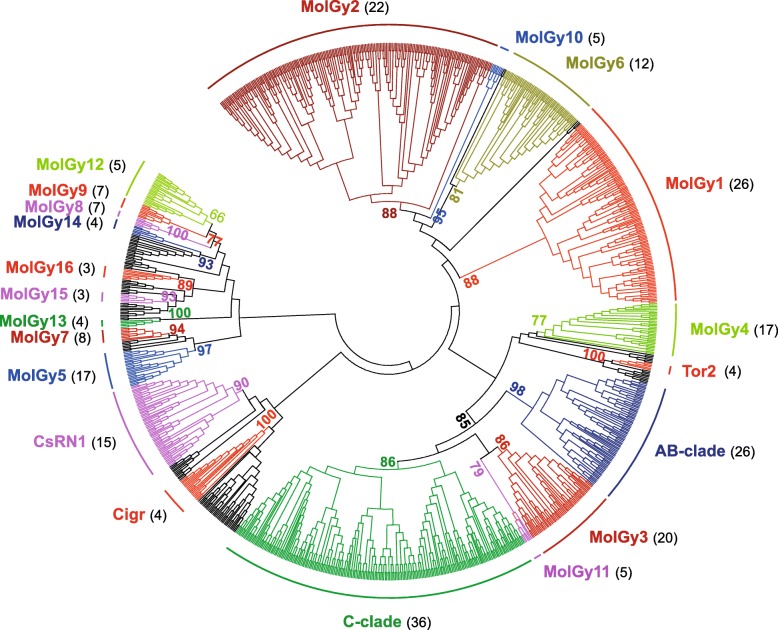
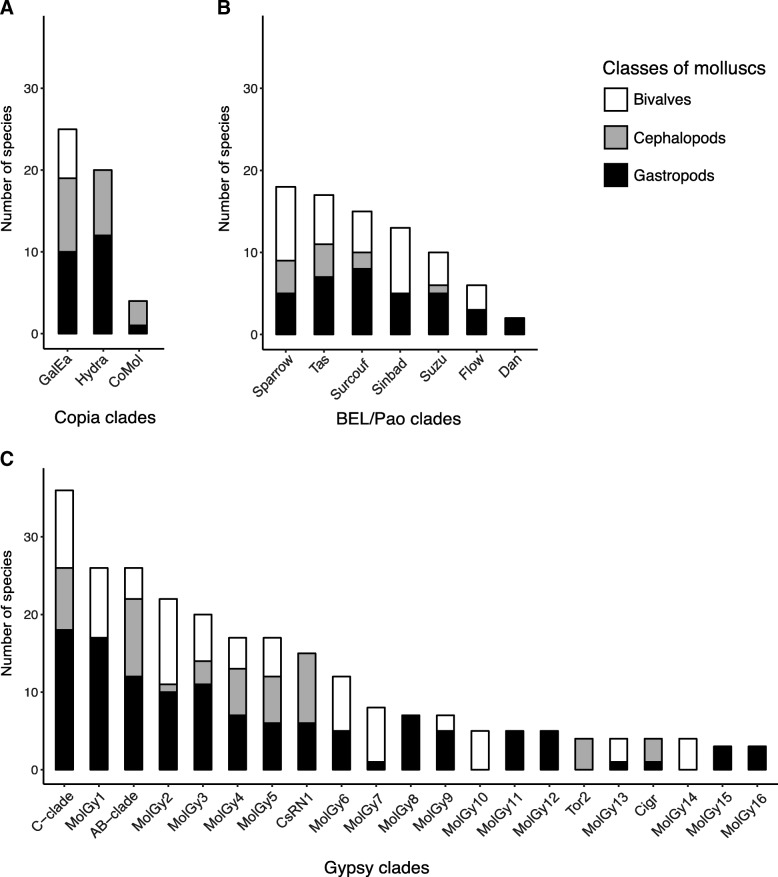
Results: We compare nine genomes of molluscs and further added LTR-retrotransposons sequences detected in databases for 47 additional species. We identified 1709 families, which enabled us to define 31 clades. We show that clade richness was highly dependent on the considered superfamily. We found only three Copia clades, including GalEa and Hydra which appear to be widely distributed and highly dominant as they account for 96% of the characterised Copia elements. Among the seven BEL/Pao clades identified, Sparrow and Surcouf are characterised for the first time. We find no BEL or Pao elements, but the rare clades Dan and Flow are present in molluscs. Finally, we characterised 21 Gypsy clades, only five of which had been previously described, the C-clade being the most abundant one. Even if they are found in the same number of host species, Copia elements are clearly less abundant than BEL/Pao elements in copy number or genomic proportions, while Gypsy elements are always the most abundant ones whatever the parameter considered.
Conclusions: Our analysis confirms the contrasting dynamics of Copia and Gypsy elements in metazoans and indicates that BEL/Pao represents the second most abundant superfamily, probably reflecting an intermediate dynamic. Altogether, the data obtained in several taxa highly suggest that these patterns can be generalised for most metazoans. Finally, we highlight the importance of using database information in complement of genome analyses when analyzing transposable element diversity.
Keywords: BEL/Pao; Comparative genomic; Copia; Gypsy; LTR- retrotransposons; Molluscs.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
